
Innovative therapies have improved the prognosis of many oncological diseases. A major limitation for the individual prognosis are disease- and therapy-associated cardiovascular complications. Even rare events can have serious consequences.
Innovative therapies have significantly improved the prognosis of many oncological diseases in recent years. A major limitation for the individual prognosis after successful cancer therapy are disease- and therapy-associated cardiovascular complications. Despite cardiotoxic effects of oncologic therapies, some of which have been known for a long time, there are few data on risk stratification and specific treatments. This is true for the classical systemic therapies (e.g. anthracyclines, taxanes or immunomodulators), but especially for the newer therapies (e.g. checkpoint inhibitors or proteasome inhibitors). Using BRAF/MEK inhibitors as an example, the therapeutic challenges from a cardio-oncology perspective will be discussed below.
Example malignant melanoma
Malignant melanoma is commonly diagnosed in both men and women and, for example, is the fourth most common manifestation of a new cancer in Switzerland, based on the 2017 Cancer League report. Mutations in oncogenes are often causally involved in the development of malignant melanoma. BRAF mutations are found in approximately half of all non-resectable or metastatic melanomas. Affected patients have had few treatment options with response rates below 20% until recently. According to the 2016 guideline, therapy with a BRAF/MEK inhibitor is therefore recommended [1].
BRAF is a cytoplasmic serine/threonine kinase that plays a regulatory role in the regulation of cell growth within the Ras-Raf-MEK1-ERK1/2 pathway. By far the most frequent mutation affects codon 600. 90% of mutations change valine to glutamic acid (V600E) [2] resulting in uncontrolled activation. This led to the development of specific inhibitors: vemurafenib and dabrafenib, as well as the downstream MEK kinase inhibitors: trametinib and cobimetinib. The combination of preparations from both substance groups was able to show a significant additional benefit with regard to progression-free survival and overall survival in clinical trials [3]. For example, in patients with untreated metastatic melanoma and BRAF V600E or V600K mutation, a median progression-free survival of 7.3 months was found with vemurafenib monotherapy compared with 11.4 months in the combination therapy group with dabrafenib and trametinib [4]. MEK inhibitors alone, as shown for trametinib, have previously been shown to improve median progression-free survival as a primary endpoint to 4.8 versus 1.5 months compared with conventional chemotherapy. A simplified overview of the inhibitors is given in Figure 1.

A number of adverse effects of BRAF and MEK inhibitors became conspicuous. Vemurafenib primarily showed arthralgia, rash, fatigue, alopecia, photosensitivity, nausea, keratoacanthoma, or squamous cell carcinoma in the phase III BRIM-3 trial [5]. Dabrafenib showed cutaneous manifestations in particular comparable to the reported adverse effects of vemurafenib, with fever seen as a more common complication in dabrafenib-treated patients [6].
Clinical data on cardiac adverse events
From a clinical point of view, on the one hand, cardiac function, depicted by left ventricular ejection fraction (LVEF), and on the other hand, newly occurring, potentially malignant arrhythmias (ventricular tachycardia, ventricular fibrillation) are relevant for the individual prognosis. Prolongation of QT time is considered an indicator of the potential occurrence of malignant arrhythmias in clinical trials.
A reduction in LVEF with combination therapy (dabrafenib and trametinib) was observed in 8% of patients, and this led to study discontinuation in 3% of patients. Half of the affected patients (approximately 4%) suffered a higher degree of LVEF limitation. There was no LVEF decrease in the vemurafenib monotherapy group. However, patients with pre-existing heart failure corresponding to NYHA II or higher were excluded from the study, so the data are not generally applicable to other collectives [4]. Rates of study discontinuation were comparable at 12% to 13% in both groups, as was overall toxicity.
In a comparable study, 2% of patients in the dabrafenib monotherapy group showed a decrease in LVEF, with 4% of patients in the combination therapy group (dabrafenib and trametinib) showing a reduction in LVEF [7]. The combination of vemurafenib and the MEK inhibitor cobimetinib also resulted in increased rates of LVEF limitations (8% vs. 3% in each group) compared with monotherapy [8]. As in the case of the described combination therapies with BRAF inhibitors, worsening of LVEF occurred in a comparable proportion of 7% with trametinib monotherapy, with 1% of patients having to discontinue the study due to the development of severely impaired LVEF [9].
Other cardiac side effects were seen in QT time prolongation (for example, in the phase II BRIM-2 trial). However, no induction of a life-threatening rhythm episode was found [10]. In larger cohorts, relevant QT prolongations greater than 480 ms were found in 7% of patients, without evidence of malignant arrhythmias. Discontinuation of vemurafenib treatment due to QT prolongation occurred in 0.5% of cases [11]. Larkin et al. also report in a large collective (3222 patients) prolonged QT times in 10% of cases, over 500 ms in 2% and two patients with relevant arrhythmias [12].
In summary, cardiac adverse events with limitations in LVEF are largely due to the addition of MEK inhibitors. The magnitude of cardiac adverse effects is comparable in MEK inhibitor therapy alone and in combination therapy with BRAF inhibitors. On the other hand, especially combination therapy is superior in terms of oncologic outcome, particularly progression-free survival and incidence of second skin malignancies.
In addition to reductions in LVEF as well as QT time prolongations, increases in blood pressure have been described for the compounds with an incidence of 15-25%. In contrast to the limitation of LVEF or the occurrence of a prolonged QT time, therapeutic measures with common antihypertensive drugs as well as clinical controls by blood pressure measurement are easy to derive in case of hypertension.
What can we learn from preclinical models ?
The molecular signaling pathways of mitogen-activating protein kinases (MAP kinases) in the cardiomyocyte have been widely studied at the cellular level and in animal models. Based on this, activation of the Ras-Raf-MEK1-ERK1/2 pathway in cardiomyocytes is associated with mediation of cellular hypertrophy and apoptosis. Members of the signaling cascade interact with a variety of other MAP kinases such as C-Jun N-terminal kinase (JNK), p38, and extracellular signal-regulated kinase 5 (ERK5) [13]. Classically, activation of ERK1/2 is associated with growth processes, whereas the MAP kinases JNK and p38 are more likely to be activated in the context of pathological stimuli. Artificial activation of the MEK1-ERK1/2 pathway is therefore more likely to be cardioprotective, whereas activation of other MAP kinases such as p38 or JNK is more likely to lead to adverse cardiac effects [14].
Mutations in the ERK1/2 pathway, namely the ERK1/2 parent phosphatase PTPN11 and the G protein K-RAS, are causally associated with congenital heart defects such as Noonan and LEOPARD syndromes. Cardiac dysrhythmias and septal defects, but most importantly hypertrophic cardiomyopathies, have been described in these syndromes.
Inactivation of PTNP11 (Y279C mutation) in mice phenocopies the cardio-facio-cutaneous syndrome including development of primary hypertrophic cardiomyopathy that transitions to dilated cardiomyopathy with advancing age of the animals [15]. In mouse models, hypertrophic cardiomyopathy could furthermore be induced by Raf1-L613V-knock-in, a human Noonan syndrome mutation [16]. In response to increased volume loading, mice showed increased interstitial and perivascular cardiac fibrosis formation and significantly reduced overall survival. MEK and ERK activity are more inducible in “knock-in” upon ligand binding; accordingly, the described cardiac phenotype could be prevented by therapy with a MEK inhibitor.
Artificial overactivation with cardiac overexpression of MEK1, on the other hand, leads to compensated cardiac hypertrophy without, at least in animal models, increased lethality. LVEF was actually improved echocardiographically with impaired diastolic function. ERK1/2 were upregulated in the MEK1 transgenic model [17]. Activation of the Raf-1/MEK/ERK pathway is also necessary for cardiac hypertrophy during increased pressure loading [18]. In pathological remodeling processes in the setting of myocardial infarction, activation of the ERK pathway also resulted in cardioprotection.
Overall, a complex picture of the benefits and adverse effects of the individual MAP kinases emerges, with a pronounced effect on cardiac hypertrophy in most papers. Consecutive changes (apoptosis, fibrosis, cardiac dysfunction) result from additional stress induction or overactivation of individual signaling pathways (Table 1).
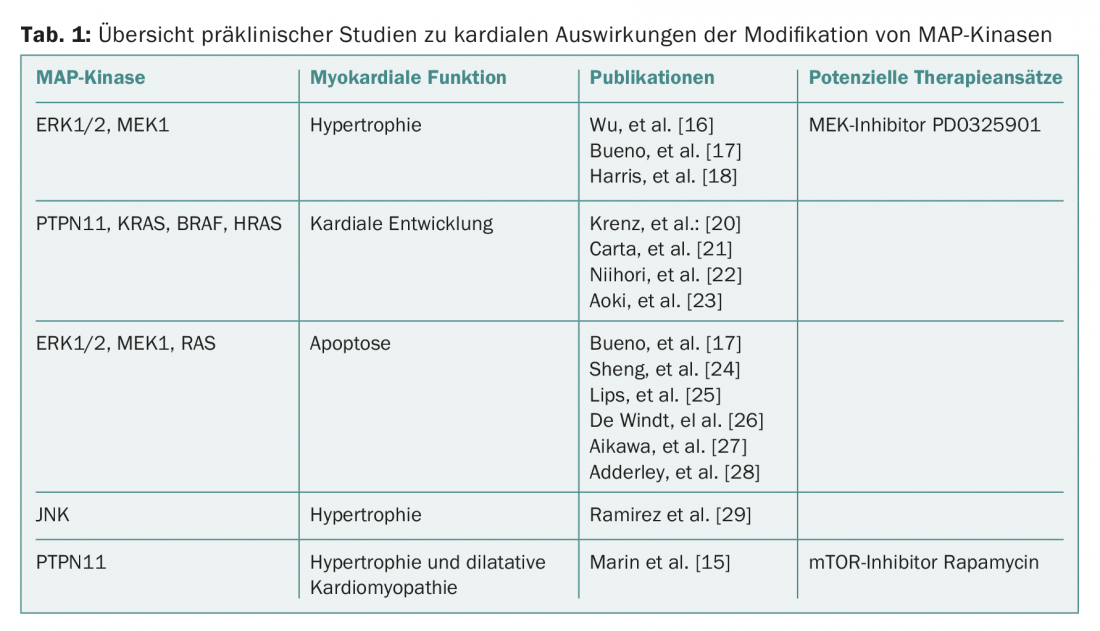
In part, the clinical observations of cardiotoxicity of MEK inhibition can be replicated. Selective intervention in the Ras-Raf-MEK-ERK1/2 pathway or affiliated signaling pathways-as shown for inhibition of mTOR with rapamycin-may be a future approach to reduce cardiac side effects in the context of oncological therapies with BRAF/MEK inhibition. It may also be suggested that state-of-the-art imaging (eg, cardiac MRI or 3D echocardiography) may identify patients with activation of prohypertrophic pathways earlier.
Conclusion for practice
Cardiac side effects of BRAF/MEK inhibitors have been documented to date in a relatively small number of patients in the single-digit percentage range. However, in case of occurrence, these may lead to serious consequences and termination of therapy. Molecular biology revealed a different picture of the individual MAP kinases, particularly with regard to cardiac hypertrophy as well as apoptosis. Most unclear are the exact cardiac stress conditions and predisposing factors in a patient that lead to clinically significant worsening of LVEF with therapy. For this reason, close and efficient cardiac monitoring of patients is necessary before and during BRAF/MEK inhibition therapy, so that affected patients (worsening of LVEF, QT time prolongation, arterial hypertension) can receive prompt cardiac therapy. If necessary, oncological therapy can then be continued under close monitoring.
According to the ESC guideline, the treatment of cardiotoxic effects consists first of all in minimizing cardiovascular risk factors, ideally before starting potentially cardiotoxic (chemo)therapy. For example, close monitoring of blood pressure may prove necessary. Preclinical work suggests a role for co-stressors as well. If LVEF is limited, heart failure therapy using beta-blockers and ACE inhibitors as first-line medications is resorted to. Preclinical data suggest that the cardiac beta-adrenergic pathway also acts via MEK and could be favorably affected with blockade. In case of a known limitation of LVEF or a high risk profile, prophylactic heart failure therapy may possibly be considered [19].
Close monitoring of cardiac biomarkers (troponin and NT-BNP) and regular ECG follow-up (especially QT time prolongation, arrhythmias) during therapy may provide early evidence of a cardiotoxic reaction. In addition, the clinical symptoms of heart failure such as edema or dyspnea must be evaluated regularly. A cardiac examination including echocardiography should be performed every 12 weeks during therapy, which may reveal early reductions in diastolic or longitudinal function or increases in pulmonary arterial pressure in addition to systolic LVEF. Based on case reports, an early onset of cardiac dysfunction within the first 14 days after initiation of therapy can be assumed for MEK inhibitors. Therefore, earlier monitoring with echocardiography and cardiac markers seems reasonable. A possible scheme for the management of high-risk patients based on therapy or predisposing factors can be found in Figure 2.
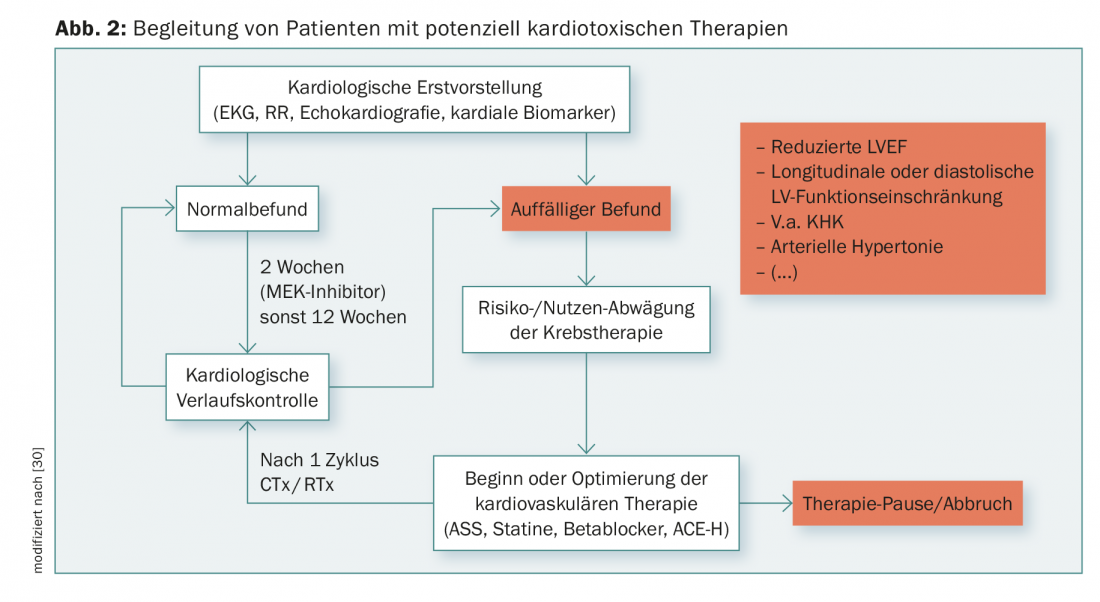
In perspective, based on molecular knowledge, specific targets of MAP kinases and affiliated signaling pathways can be identified, enabling efficient treatment of cardiac side effects.
Take-Home Messages
- Cardiac side effects of BRAF/MEK inhibition have so far been in the single-digit percentage range. However, when they do occur, they can lead to serious consequences and therapy discontinuation.
- According to the ESC, treatment of cardiotoxic effects consists first of all in minimizing cardiovascular risk factors – ideally before starting potentially cardiotoxic therapy.
- Thereafter, close monitoring of cardiac biomarkers as well as regular ECG follow-up and assessment of clinical symptoms during therapy may provide early evidence of a cardiotoxic response.
- An interdisciplinary approach by the treating oncologists and cardiologists improves the individual cardiac prognosis.
Literature:
- Dummer R, et al: The updated Swiss guidelines 2016 for the treatment and follow-up of cutaneous melanoma. Swiss Med Wkly 2016; 146: w14279.
- Cheng L, et al: Molecular testing for BRAF mutations to inform melanoma treatment decisions: a move toward precision medicine. Mod Pathol 2018; 31(1): 24-38.
- Long GV, et al: Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. Lancet 2015; 386(9992): 444-451.
- Robert C, et al: Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med 2015; 372(1): 30-39.
- Chapman PB, et al: Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011; 364(26): 2507-2516.
- Hauschild A, et al: Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012; 380(9839): 358-365.
- Long GV, et al: Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med 2014; 371(20): 1877-1888.
- Larkin J, et al: Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med 2014; 371(20): 1867-1876.
- Flaherty KT, et al: Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med 2012; 367(2): 107-114.
- Ribas A, et al: BRIM-2: An open-label, multicenter phase II study of vemurafenib in previously treated patients with BRAF V600E mutation-positive metastatic melanoma. Journal of Clinical Oncology 2011; 29(15 suppl): 8509-8509.
- Flaherty L, et al: A single-arm, open-label, expanded access study of vemurafenib in patients with metastatic melanoma in the United States. Cancer J 2014; 20(1): 18-24.
- Larkin J, et al: Vemurafenib in patients with BRAF(V600) mutated metastatic melanoma: an open-label, multicentre, safety study. Lancet Oncol 2014; 15(4): 436-444.
- Wang Y: Mitogen-activated protein kinases in heart development and diseases. Circulation 2007; 116(12): 1413-1423.
- Molkentin JD: Parsing good versus bad signaling pathways in the heart: role of calcineurin-nuclear factor of activated T-cells. Circ Res 2013; 113(1): 16-19.
- Marin TM, et al: Rapamycin reverses hypertrophic cardiomyopathy in a mouse model of LEOPARD syndrome-associated PTPN11 mutation. J Clin Invest 2011; 121(3): 1026-1043.
- Wu X, et al: MEK-ERK pathway modulation ameliorates disease phenotypes in a mouse model of Noonan syndrome associated with the Raf1(L613V) mutation. J Clin Invest 2011; 121(3): 1009-1025.
- Bueno OF, et al: The MEK1-ERK1/2 signaling pathway promotes compensated cardiac hypertrophy in transgenic mice. EMBO J 2000; 19(23): 6341-6350.
- Harris IS, et al: Raf-1 kinase is required for cardiac hypertrophy and cardiomyocyte survival in response to pressure overload. Circulation 2004; 110(6): 718-723.
- Zamorano JL, et al: 2016 ESC Position Paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC Committee for Practice Guidelines: The Task Force for cancer treatments and cardiovascular toxicity of the European Society of Cardiology (ESC). Eur Heart J 2016; 37(36): 2768-2801.
- Krenz M, Yutzey KE, Robbins J: Noonan syndrome mutation Q79R in Shp2 increases proliferation of valve primordia mesenchymal cells via extracellular signal-regulated kinase 1/2 signaling. Circ Res 2005; 97(8): 813-820.
- Carta C, et al: Germline missense mutations affecting KRAS isoform B are associated with a severe Noonan syndrome phenotype. Am J Hum Genet 2006; 79(1): 129-135.
- Niihori T, et al: Germline KRAS and BRAF mutations in cardio-facio-cutaneous syndrome. Nat Genet 2006; 38(3): 294-296.
- Aoki Y, et al: Germline mutations in HRAS proto-oncogene cause Costello syndrome. Nat Genet 2005; 37(10): 1038-1040.
- Sheng Z, et al: Cardiotrophin 1 (CT-1) inhibition of cardiac myocyte apoptosis via a mitogen-activated protein kinase-dependent pathway. Divergence from downstream CT-1 signals for myocardial cell hypertrophy. J Biol Chem 1997; 272(9): 5783-5791.
- Lips DJ, et al: MEK1-ERK2 signaling pathway protects myocardium from ischemic injury in vivo. Circulation 2004; 109(16): 1938-1941.
- De Windt LJ, et al: Calcineurin-mediated hypertrophy protects cardiomyocytes from apoptosis in vitro and in vivo: An apoptosis-independent model of dilated heart failure. Circ Res 2000; 86(3): 255-263.
- Aikawa R, et al: Oxidative stress activates extracellular signal-regulated kinases through Src and Ras in cultured cardiac myocytes of neonatal rats. J Clin Invest 1997; 100(7): 1813-1821.
- Adderley SR, Fitzgerald DJ: Oxidative damage of cardiomyocytes is limited by extracellular regulated kinases 1/2-mediated induction of cyclooxygenase-2. J Biol Chem 1999; 274(8): 5038-5046.
- Ramirez MT, et al: The MEKK-JNK pathway is stimulated by alpha1-adrenergic receptor and ras activation and is associated with in vitro and in vivo cardiac hypertrophy. J Biol Chem 1997; 272(22): 14057-14061.
- Tilemann LM, et al: Cardio-oncology: conflicting priorities of anticancer treatment and cardiovascular outcome. Clin Res Cardiol 2018; 107(4): 271-280.
InFo ONCOLOGY & HEMATOLOGY 2018; 6(4): 8-12.









