Interstitial lung diseases are frequently encountered by primary care physicians, especially in their early forms. The differentiation of the various forms is often not obvious at first sight and requires a precise diagnosis with anamnesis. An expert from the University Hospital Basel provided an overview of the interstitial pneumopathies that should be considered.
Pulmonary fibrosis is a relatively rare disease associated with compaction of the lung parenchyma. This change results in a decrease in lung volume and an obstruction to oxygen uptake. Prognostically, fibrotic remodeling of the lung is considered unfavorable, stated Dr. Kathleen Jahn of the Department of Pneumology at University Hospital Basel.
Idiopathic pulmonary fibrosis
Idiopathic pulmonary fibrosis has a very unfavorable prognosis with a median survival of 1.5 to 3 years after initial diagnosis. Nevertheless, fulminant courses, relapses, but also comparatively slow progressions can occur.
Pathophysiologically, we are dealing with an inflammatory response that involves the release of cytokines and the accumulation of inflammatory products. Oxidative stress increases, leading to compaction of the extracellular matrix. Fibroblasts are activated, and growth hormone dysregulation occurs in lung tissue.
In lung function, this presents as shrinkage of the lungs. “It gets smaller and tighter and can’t expand as well. In clinical practice, you see that the thorax can’t move as well with advanced pulmonary fibrosis, there are restrictions,” Dr. Jahn explained. The one-second capacity (peak flow) may not necessarily be restricted, but the total volume shrinks overall and the lungs can no longer fill completely.
Respiratory bronchiolitis (RB-ILD).
RB-ILD is the most benign of the interstitial pneumopathies. Patients typically have a smoking history that has been persistent for many years (min. 20 PY), and most are also not looking at a long-lasting nicotine cessation when they present with bronchiolitis. Complaints are nonspecific: cough (productive and nonproductive) and difficulty breathing on exertion, auscultatory usually rales. CT shows a centrinodular pattern (OL-emphasized), small nodules (<5 mm) are found throughout the lung parenchyma, some of which are surrounded by milky glass, there may be incipient bronchial wall thickening. Classical fibrotic remodeling is very rarely seen in RB-ILD.
The gold standard in diagnostics is bronchoscopy with lavage (BAL). Smoker’s macrophages are the most visible. Less common and usually unnecessary are transbronchial biopsies (TBB), which image bronchiolocentric anthracosis deposits and neutrophilic inflammation. The therapy is relatively simple: first and foremost, stop smoking. Two-thirds of cases show clinical improvement if they can achieve consistent smoking cessation. Prednisone (initial 0.5 mg/kgKG) is rarely needed and only when the presentation is advanced.
Sarcoidosis
The mean age of manifestation of sarcoidosis is between 20 and 60 years. Again, the symptoms are nonspecific: nonproductive cough, performance intolerance, chest pain, and fever are among them, as are unclear constellations of infections. Less commonly, weight loss and fatigue may also occur.
Imaging (CT chest) shows bihilar mediastinal lymphadenopathy in 50%, milk glass nodules may also be seen, and in advanced stages with chronic inflammation, parenchymal destruction and fibrotic changes may also occur. Additively, PET-CT may be useful to demonstrate lymphadenopathy and exclude differential diagnoses of a malignant nature. In the early stages, the diagnosis can usually only be manifested by spiroergometry. Under stress, a relevant decrease in pO2 is observed, which may be an early sign of sarcoidosis.
Bronchoscopy is again the gold standard for confirming the diagnosis. Moderate lymphocytosis (approximately 20-30%) is found with increased CD4/CD8 quotient >2 in BAL. A cobblestone-like relief suggests granulomatous inflammation (SH-Bx); biopsy sampling (TBB) reveals productive, noncaseating granulomas. The therapy is determined individually depending on the organ involvement and the clinic:
asymptomatic patients and those with negative predictive markers do not require therapy.
Indicators for therapy are hypercalcemia/hypercalciuria, occult involvement, and diffusion restrictions.
Usually, one starts with monotherapy prednisone 0.3-0.6 mg/kgKG (approx, 20-40 mg/day). This is tapered in 5-10 mg increments every 2-4 weeks. The initial duration of therapy should be approximately 3 to 6 months. If this does not lead to the desired improvement, additive azathioprine or mycophenolate can be used. This mostly affects patients with type 2 diabetes or osteoporosis. As always with chronic steroid therapy, it should also be remembered to establish gastric protection (PPI) and perform pneumocystis prophylaxis (3×/week), the expert reminded. It is also very important not to add calcium in sarcoidosis, as this leads to macrophage activation.
OP and COP
Organizing pneumonia (OP) and cryptogenic organizing pneumonia (COP) look completely the same on CT. Infectious, classic causes are usually present during surgery. “On the other hand, we refer to as cryptogenically organizing only those pneumonias that have no clear trigger and are then considered idiopathic,” Dr. Jahn said.
In OP and COP, alveolar epithelial damage is present, and fibrin exudation into the alveolar space occurs. So-called migrating infiltrates are typical for COP. Therapeutically, surgery can usually be performed with a watch-and-wait strategy regimen if functional limitation is only mild. Prednisone (PDN) is again indicated for more severe courses. “You see: Cortisone is a relative wonder drug in the interstitial pneumopathies.” However, due to a relapse rate of around 30%, one should tap down slowly here as well.
If there is a relapse, higher dose steroids should be started again. Possible differential diagnoses in cases of frequent recurrences despite good response to established PDN therapy include immunodeficiency syndrome, granulomatosis with polyangitis and other rheumatologic diseases, chronic thyroiditis, and also chronic inflammatory bowel disease.
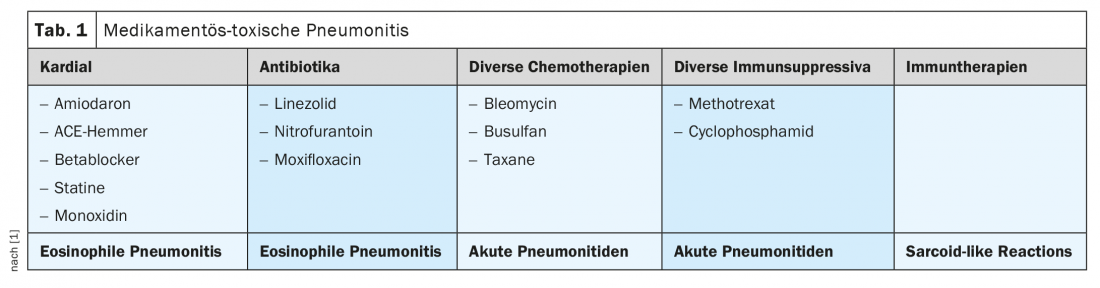
Drug toxic pneumonitis
In the case of drug-toxic pneumonitis (Tab. 1) with various triggers, BAL is usually useful in confirming the diagnosis; TBB is rarely necessary. Therapeutically, stopping the causative agent is indicated; depending on the clinic, supportive PDN therapy may be appropriate.
Idiopathic pulmonary fibrosis (IPF)
Most interstitial pneumopathies respond very well to steroids – IPF, on the other hand, is completely steroid resistant. However, targeted therapeutic options have been available for several years.
In IPF, there is often a familial clustering, especially if the affected person is younger than 55 years of age. The classic picture of a restriction is seen, the volumes become smaller, there are hyperventilation patterns. In the laboratory, a rheumatic constellation should be excluded as the underlying disease. The fewer of the typical changes seen on CT imaging, the less likely IPF is, the expert explained.
The two active ingredients pirfenidone and nintedanib can be used for treatment. Both have anti-fibrotic, anti-proliferative and anti-inflammatory effects. Both have been tested in large studies and have been on the market for several years. They both reduce the decline in lung volume. But: They do not cause a reventable process – what fibrosis is present cannot be reduced again. In addition, both drugs are relatively poorly tolerated and cause abdominal discomfort, nausea, diarrhea, and dyspepsia, among other symptoms. Pirfenidone causes high phototoxicity – so sun-loving patients should be adjusted to nintedanib rather than pirfenidone, Dr. Jahn advised. Nintedanib, however, leads to more hepatopathies on the other side. It is also contraindicated when taking oral anticoagulants due to an increased risk of bleeding.
Take-Home Messages
- Early functional site assessment and functional clarification in progressive dyspnea.
- Comprehensive history regarding familiality, exposure toxins, polymedication
- Tight controls
- Many ILDs are steroid-responsive and show a good response
- IPF is a special case, as NOT steroid-responsive, but delayable in progression with anti-proliferative agents.
- BAL usually remains nonspecific.
- Diagnosis by lung biopsy (endoscopic or surgical) indicated
- Attention to drug side effects
Source:
- Jahn K: Common interstitial lung diseases – diagnosis and therapy. FomF Update Refresher General Internal Medicine, 25.1.2022 (virtual)
HAUSARZT PRAXIS 2022; 17(5): 43-44
InFo PNEUMOLOGY & ALLERGOLOGY 2022; 4(2): 38-39.