H1 antihistamines are divided into a first, older generation with sedative side effects and a 2nd generation without these effects. Our author sheds light on the pharmacology of this group of agents.
So far, four different histamine receptors are known. These G-protein-coupled molecules are located on the cell surface and exert different effects depending on the site of expression. While activation of the H1 receptor leads in particular to pruritus, vasodilation, smooth muscle contraction with bronchospasm or abdominal cramps, mucus secretion with rhinorrhea and an increase in bronchial secretions as well as an increase in vascular permeability, H2 receptors are involved in particular in increasing gastric juice and acid secretion. In addition, there are H3 receptors, which play a role in the CNS as presynaptic autoreceptors, and H4 receptors, which play a role in the differentiation and modulation of immune cells, among others. In this article, the substances directed against the H1 receptor are discussed, whereas H2 receptor antagonists, of which only ranitidine is still on the market in Switzerland, are not. Agonists and antagonists at the H3 and H4 receptors are in clinical development.
Pharmacodynamics of H1 antihistamines.
In Switzerland, 22 active substances of the H1 antihistamine class are available. It is now thought that antihistamines stabilize the H1 receptor in its inactive conformation, ensuring that fewer receptors can be activated by histamine. While H1 antihistamines of the first, older generation penetrate well into the central nervous system and exert a sedative effect there at postsynaptic H1 receptors, this is not the case with the representatives of the 2nd generation or not in therapeutic concentrations (Tab. 1) . Due to their good efficacy at central H1 receptors, some representatives of the 1st generation used as sedatives/hypnotics (e.g. doxylamine, diphenhydramine), antiemetics (e.g. meclozine) or for motion sickness (e.g. dimenhydrinate). The poor CNS penetration of the representatives of the 2nd generation is due to the fact that these substances are hydrophilic and substrates of the outwardly directed transporter P-glycoprotein present in the blood-brain barrier (among other membrane barriers of the body). This prevents sedation, which is used in anti-allergic indications with substances of the 1st generation was often therapy-limiting. Some H1 antihistamines of the 1st generation have additional effects on acetylcholine, norepinephrine and serotonin receptors, while representatives of the 2nd generation specifically inactivate the H1 receptor.
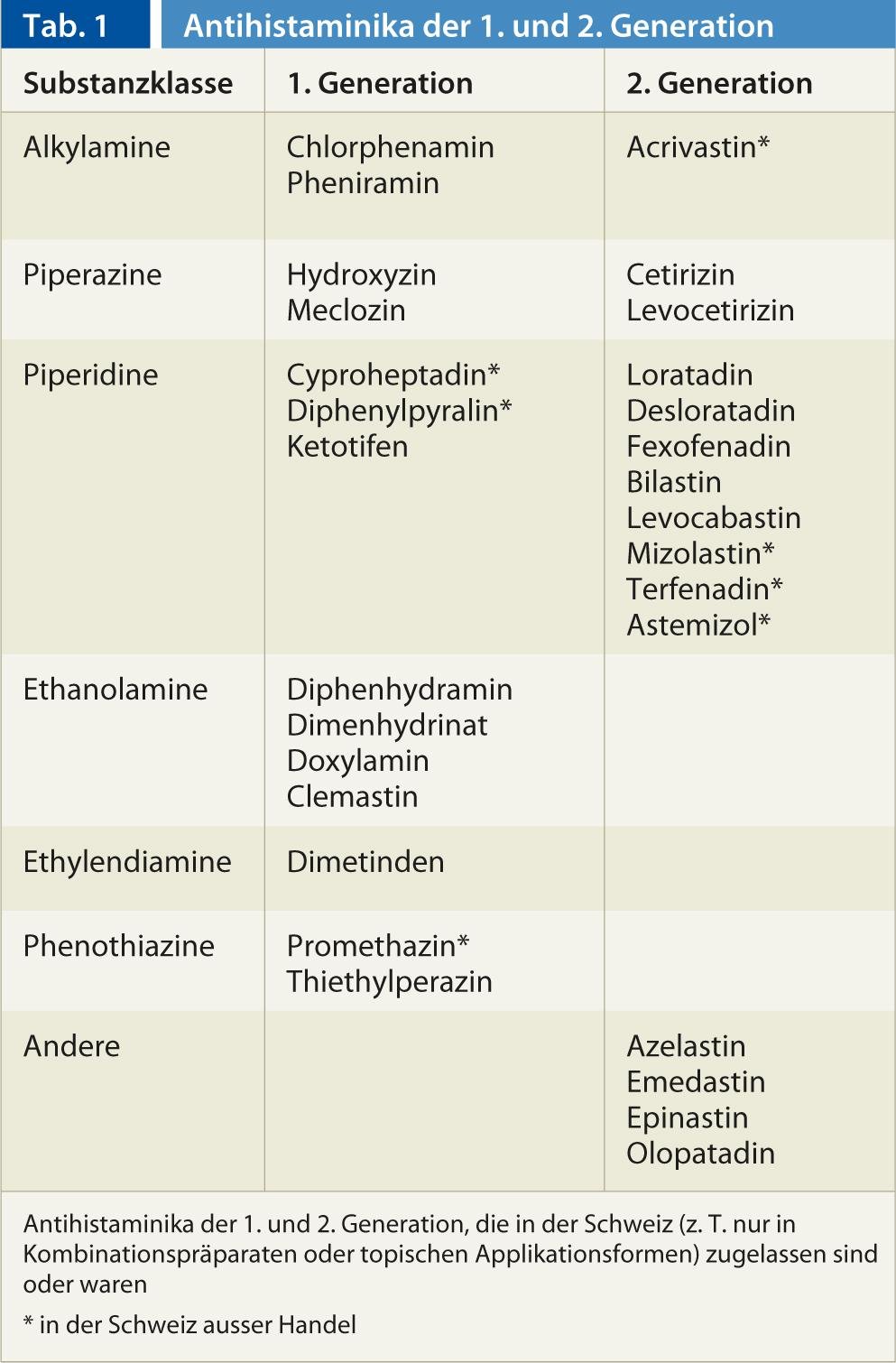
Overall, the clinical efficacy of H1 antihistamines of the 1st generation have been poorly studied in clinical trials, while the evidence for the use of H1 antihistamines of the 2nd generation is good for allergic rhinitis, allergic conjunctivitis and urticaria. The use of products from the 2nd generation in atopic dermatitis, asthma, anaphylaxis, non-allergic angioedema, colds, itching of non-allergic origin, etc. has been poorly studied or the studies did not show convincing effects and there is also no approval for such indications. In children, substances of the 1st generation can lead to sometimes threatening side effects, so that the indication must be made with particular care. In a so-called 3rd generation, enantiomers or metabolites of molecules of the 2nd generation, with no major pharmacodynamic differences, so that the substances are pharmacologically similar to the 2nd generation.
Pharmacokinetics of H1 antihistamines.
The first hurdle that a drug has to overcome in order to be effective is taking it. Ensuring compliance or adherence is therefore of particular importance in everyday medical practice. Most H1 antihistamines for systemic use are available as solid dosage forms (tablets, dragées, film-coated tablets, suppositories) or drops for oral administration. Some substances are applied locally (e.g. eye drops). Only a few can also be administered intravenously (dimetinden, clemastine, thiethylperazine). Absorption, which is rapid for most 2nd generation H1 antihistamines and results in peak levels after one to three hours, is followed by distribution in blood and tissues, metabolism if appropriate, and excretion.
In particular, there are large differences in metabolism and elimination between antihistamines (Table 2). Furthermore, there are differences in metabolism between people caused by genetic and environmental influences. In particular, the cytochrome P450 enzyme CYP2D6, which is involved in the degradation of some H1 antihistamines (Table 2), shows strong genetic variability that can range from absence to multiplication of normal enzyme activity. In addition, CYP2D6 can be inhibited by some substances: bupropion, cinacalcet, the selective serotonin reuptake inhibitors paroxetine, fluoxetine and duloxetine, and the antifungal drug terbinafine are among the more potent CYP2D6 inhibitors. However, because all H1 antihistamines are cleared by more than one enzyme and are often renally eliminated unchanged, genetic variability and inhibition of CYP2D6 very rarely play a role in the use of H1 antihistamines.
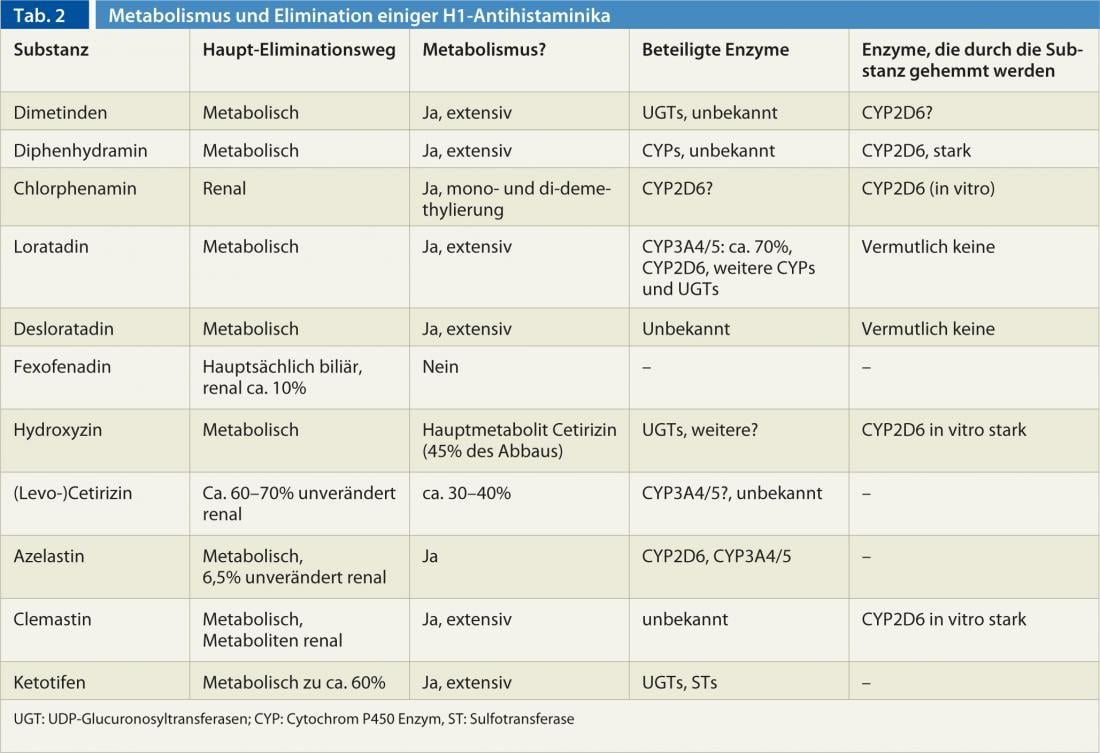
Some 1st generation H1 antihistamines are also CYP2D6 inhibitors. (Tab. 2), so that when CYP2D6 substrates are used concomitantly (e.g., codeine, dextromethorphan, many antipsychotics [haloperidol, risperidone, aripiprazole], atomoxetine, many antidepressants [most tricyclics, venlafaxine, and others], and the beta-blockers metoprolol, carvedilol, and timolol), a lower dose of the substrate should be selected to avoid adverse effects.
Overall, the pharmacokinetics of the substances of the 1st generation are only incompletely known. generation is only incompletely known, as the substances were approved decades ago with significantly less data than is necessary today. However, there are also gaps in our knowledge of 2nd generation preparations: Desloratadine is metabolized to the equally active metabolite 3-hydroxy-desloratadine, but the enzyme or enzymes involved are unknown, although it has been found that 2% of Europeans and up to 20% of Africans cannot form 3-hydroxy-desloratadine. The CYP2D6 and CYP3A4/5 enzymes involved in loratadine metabolism do not appear to be responsible for this.
CYP3A4 is the most important cytochrome P450 enzyme both in terms of quantity and number of substrates. While function-altering genetic variants are not known (despite intensive research) for CYP3A4, there are drugs that increase enzyme activity (rifampicin, efavirenz, phenytoin, carbamazepine, St. John’s wort ingredients, et al.(azole antifungals, erythromycin, clarithromycin, ritonavir, verapamil, diltiazem, amiodarone, grapefruit ingredients, especially in grapefruit juice, etc.). CYP3A5, on the other hand, which degrades essentially the same drugs as CYP3A4, is almost never present in Europeans due to a genetic variant, whereas it is usually functional in Africans. However, except for loratadine, azelastine, and presumably cetirizine, CYP3A4/5 does not play a role in antihistamines.
It was all the more surprising when an effect of grapefruit juice was demonstrated in a study with fexofenadine: When fexofenadine was taken together with grapefruit juice, fexofenadine levels decreased, especially shortly after ingestion, compared with ingestion with water (an increase would have been expected if CYP3A4 was inhibited by the furanocoumarins of grapefruit juice). This effect can be explained by the fact that other substances in grapefruit juice (the flavonoid naringin) inhibit the intestinal inward transporter OATP1A2, which is necessary for the uptake of fexofenadine from the intestinal lumen.
Another interaction that has not yet been fully elucidated also involves fexofenadine: concomitant administration of itraconazole resulted in many times higher fexofenadine levels. Since fexofenadine is not metabolized by CYP3A, which is potently inhibited by itraconazole, the interaction was explained by inhibition of P-glycoprotein, the transporter thought to play a major role in fexofenadine elimination. Whether this hypothesis is correct and other azole antifungals also cause level elevations remains to be investigated.
On the other hand, it should be noted that substances such as (levo-)cetirizine are mainly eliminated renally, so that restrictions in renal function must also lead to dose reductions. For levocetirizine, for example, a dose reduction is already required from a glomerular filtration rate of less than 50 mL/min, an administration of 5 mg every 2nd day is prescribed; in the case of more severe renal function impairment, the dosing interval must be extended even further.
In Switzerland, patient-specific information on side effects and interactions, dose adjustments and other drug-related problems can be obtained from the clinical pharmacology facilities at university hospitals.
Adverse effects of antihistamines: Focus QTc time.
While most of the adverse effects of the H1 antihistamines are due to their action at the H1 receptor (fatigue, decreased cognitive and psychomotor performance, increased appetite) or (for the older compounds) by effects at the m-acetylcholine receptor (dry mouth, urinary retention, tachycardia), at the alpha-adrenoceptor (hypotension, dizziness, reflex tachycardia), or at the serotonin receptor (eg. increased appetite), it is less well known that some H1 antihistamines also inhibit cardiac ion channels, in particular the IKr channel, which modulates the rapid outflow of potassium during repolarization and can thus lead to a prolongation of repolarization (of the QT interval in the ECG) up to torsades de pointes ventricular tachycardia.
Overall, the potential of H1 antihistamines to prolong QTc time on ECG is poorly studied. The interest in this potentially fatal side effect was aroused when several reports of torsades de pointes episodes were available for the first 2nd generation H1 antihistamines, terfenadine and astemizole. In most cases, overdoses had been taken or concentration-increasing interactions had not been observed. The two compounds were withdrawn from the market in 1990 due to ventricular arrhythmias. For loratadine, fexofenadine, and cetirizine, there are single-case reports of prolongation of QTc time and, in some cases, torsades de pointes tachycardia.
General risk factors for QTc time prolongation are shown in Table 3 . Therefore, it seems advisable, especially in high-risk patients under regular antihistamine administration and at high doses, to obtain an ECG and to check serum potassium and magnesium.

CONCLUSION FOR PRACTICE
- 2nd generation H1 antihistamines are generally well tolerated.
- QTc time prolongation is possible, especially with overdoses and risk factors.
- Grapefruit juice should not be drunk together with fexofenadine as it decreases fexofenadine concentrations. Itraconazole increases fexofenadine concentrations, which may lead to symptoms of overdose.
- For mainly renally excreted antihistamines such as (levo-)cetirizine, the dose must be adjusted in renal insufficiency.
PD Alexander Jetter, MD
Literature:
- Simons FE, Simons KJ: Histamine and H1-antihistamines: celebrating a century of progress. J Allergy Clin Immunol 2011; 128: 1139-1150.
- Shon JH, Yoon YR, Hong WS, Nguyen PM, Lee SS, Choi YG, Cha IJ, Shin JG: Effect of itraconazole on the pharmacokinetics and pharmacodynamics of fexofenadine in relation to the MDR1 genetic polymorphism. Clin Pharmacol Ther 2005; 78: 191-201.
- Banfield C, Gupta S, Marino M, Lim J, Affrime M: Grapefruit juice reduces the oral bioavailability of fexofenadine but not desloratadine. Clin Pharmacokinet 2002; 41: 311-318.
- Hondeghem LM, Dujardin K, Hoffmann P, Dumotier B, De Clerck F: Drug-induced QTc prolongation dangerously underestimates proarrhythmic potential: lessons from terfenadine. J Cardiovasc Pharmacol 2011; 57: 589-597.
- Compalati E, Baena-Cagnani R, Penagos M, Badellino H, Braido F, Gómez RM, Canonica GW, Baena-Cagnani CE: Systematic review on the efficacy of fexofenadine in seasonal allergic rhinitis: a meta-analysis of randomized, double-blind, placebo-controlled clinical trials. Int Arch Allergy Immunol 2011; 156: 1-15.
HAUSARZT PRAXIS 2013; 8(3): 14-17