
Adults with spinal muscular atrophy form a clinically and genetically very heterogeneous group. A common feature is the progressive loss of motor anterior horn cells in the spinal cord, resulting in impaired impulse transmission. Muscle atrophy, muscle hypotonia and paresis may result. Therefore, the therapy should individually consider the disease activity and needs of the patient.
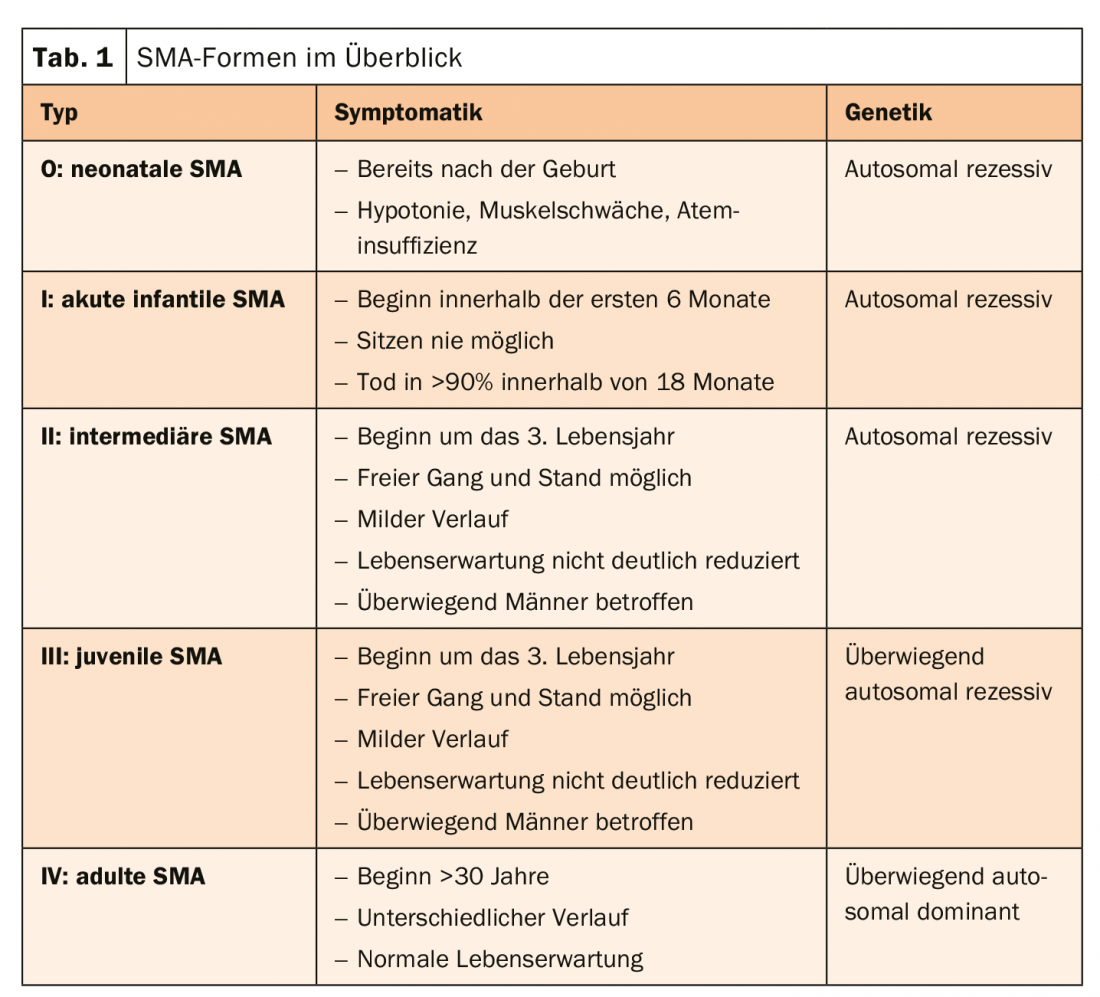
Spinal muscular atrophy (SMA) is a rare, predominantly autosomal recessive muscle disease in which motor anterior horn cells and motor cranial nerve nuclei degenerate. The cause is usually a defect in the SMN1 gene. The resulting lack of functional SMN protein can be partially compensated by the SMN2 gene. Therefore, the more SMN2 copies are present, the later the onset of SMA and the more favorable the course. Patients with late-onset SMA (types II-IV) often have a normal life expectancy. Basically, however, the disease forms a very heterogeneous group with numerous, even very rare forms. They are differentiated according to distribution pattern, onset of disease, severity, and pattern of inheritance. However, the vast majority (90%) is proximal SMA, which is characterized by the onset of muscle weakness in proximal muscle groups (types I-III). Type IV is a special form of the non-proximal type with an onset of disease >30 years (Table 1).
Individual treatment goals
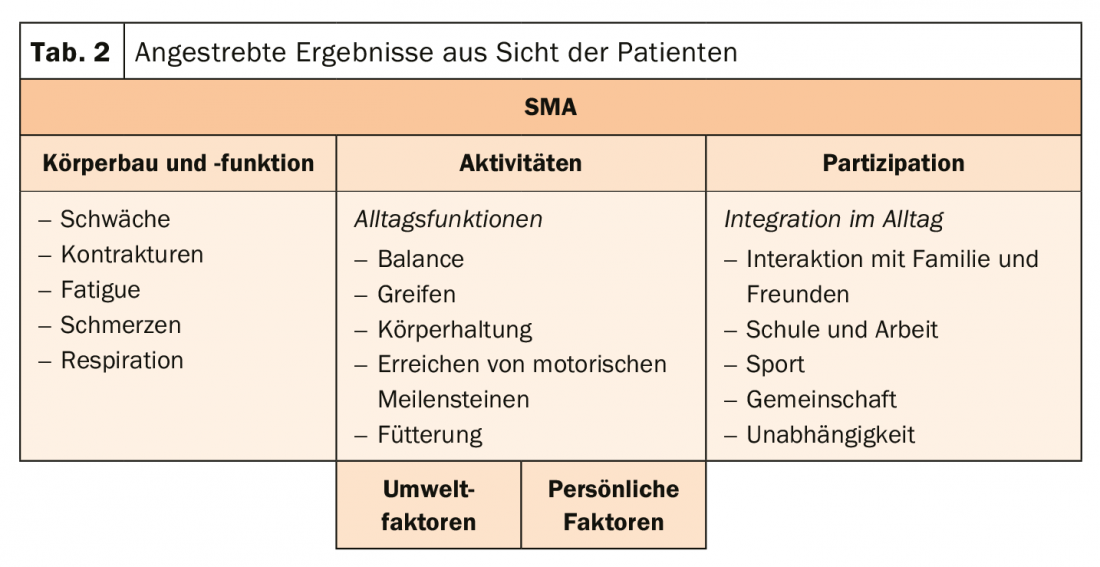
Because the group of adult patients with SMA is so diverse, treatment goals cannot be lumped together. For one person, it is important to maintain walking independence and delay the time of needing to use a wheelchair as much as possible. In other patients, however, the focus should be on the function of the upper extremities (tab. 2) . The SMA patients therefore receive physiotherapy, supportive mechanical ventilation if desired, and orthopedic measures to compensate for the curved spine. Also, care should be taken to ensure adequate caloric intake, so that the muscles do not break down even faster. Basically, treatment management aims to strengthen endurance and resistance, maintain bone health, prevent fractures, keep body weight constant, and monitor breathing.
No longer supportive-palliative treatment alone
The first disease-modifying therapy is now available. Since the pivotal studies were conducted in children, an observational study was now conducted to look more closely at the effect in adults. The results suggest that the antisense oligonucleotide nusinersen can also significantly improve motor function in adults with SMA. The change in the Hammersmith Functional Motor Scale Expanded (HFMSE) was assessed. Compared to baseline, this was significantly increased.
Source: EAN Virtual Congress 2020
InFo NEUROLOGY & PSYCHIATRY 2020; 18(4): 36 (published 6/29/20, ahead of print).









