
In addition to standard care for all forms of ALS, there has been significant progress, particularly for monogenetic forms of ALS, thanks to the development of specific gene-based treatment methods, currently ASO in particular. Basic genetic diagnostics, at least of the most common genes (SOD1, C9orf72, FUS, TARDP), are therefore recommended for all ALS patients at the time of diagnosis.
You can take the CME test in our learning platform after recommended review of the materials. Please click on the following button:
Cases of amyotrophic lateral sclerosis (ALS) were first described by Jean Martin Charcot in 1873 [1]. Charcot already provided neuropathological evidence of an underlying motor system degeneration. It is now known that amyotrophic lateral sclerosis, despite the predominant degeneration of the first and second motor neurons and the corticospinal tract, is a multisystemic neurodegeneration with numerous extramotor manifestations. We have gained increasing knowledge about the underlying genetic factors, particularly in the last decade, which has also led to the first direct therapeutic consequences.
Epidemiology
Based on data from the best-managed patient registry in Swabia, it can be estimated that there are around 8,000 to 9,000 people affected by ALS in Germany as a whole [2]. With an average age of onset of 70 to 75 years and a slight male predominance, an incidence of approx. 3/100,000 patients is assumed. The lifetime prevalence as the most descriptive statistical measure for the probability of developing ALS is 1:400. These epidemiological figures are highly congruent with figures from other European countries. In other parts of the world, such as Asia, the epidemiological data is different [3,4].
Clinical symptoms
The core symptoms of ALS are focal onset, progressive, atrophic paresis with frequent muscle spasms and fasciculations as a sign of increasing damage to the second motor neurons at the spinal level or in the brainstem area [5]. This is preceded or accompanied by an affection of the first motor neurons in the primary motor cortex and the corticospinal tract with increased and skipping muscle stretch reflexes and evidence of pathological reflexes or an increase in muscle tone in the sense of spasticity.
Clinically and in everyday life relevant cognitive deficits and behavioral abnormalities in the sense of a frontotemporal dementia syndrome are found as extra-motor symptoms in approx. 5% of people with ALS [6,7]. Accompanying disorders of the autonomic nervous system have been increasingly described in recent years [8]. In addition, pain of various origins is also important during the course of the disease [9].
Diagnostic procedures and diagnosis
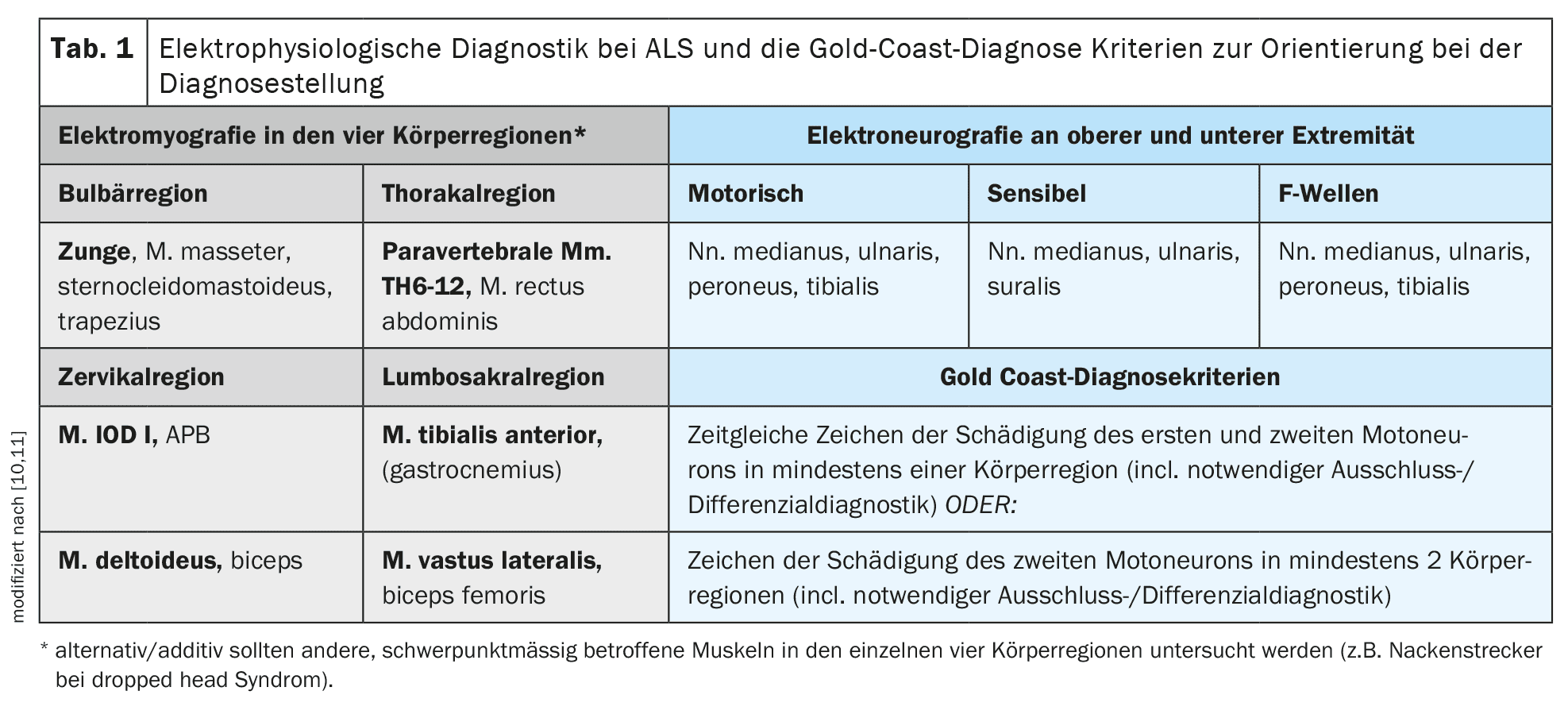
The diagnosis is primarily made clinically after consistent and careful exclusion of alternative differential diagnoses. The Gold Coast criteria [10] provide good guidance for clinical diagnosis. The most important supporting diagnostic procedure is electromyography for the detection of active denervation or polytopic fasciculation potentials with simultaneous evidence of chronic neurogenic damage. Electromyography should examine different muscles in all four regions of the body (cranial nerves/bulbar – cervical – thoracic – lumbosacral) [11]. In addition to electromyography, muscle sonography can also be performed to determine the presence of polytopic muscle fasciculations and to assess the muscular trophism and internal structure. Muscle sonography can be particularly advantageous for detecting muscle fasciculations in larger or deeper muscles, such as parts of the quadriceps femoris muscle or tongue base muscles, and can be an important complement to electromyography. Electroneurography of motor and sensory nerves as well as F-wave diagnostics on the upper and lower extremities is particularly necessary to rule out primary demyelinating neuropathy and, in particular, conduction blocks (CIDP or multifocal motor neuropathy/MMN).
Transcranial magnetic stimulation is available for recording motor-evoked potentials to objectify and, if necessary, quantify involvement of the first motor neurons. This examination method can be used to examine the central motor conduction of the thickly myelinated axons of the corticospinal tract from the motor cortex as a stimulation site along the entire spinal cord [12]. It makes sense to examine one distal muscle each on the upper and lower extremities. Table 1 provides a detailed overview of the proposed electrophysiological diagnostics for ALS and the Gold Coast diagnostic criteria .
Neurofilament light chains (NfL) are an important biomarker [13]. These can be easily determined in serum and cerebrospinal fluid. It is important that the determination is carried out using a sufficiently sensitive detection method in an established laboratory with age-adjusted limit values. It should be emphasized that although NfL can represent an additional important component for the diagnosis, an NfL within the normal range cannot rule out the diagnosis of ALS and elevated NfL values can also occur in other differential diagnoses of ALS (CIDP, vasculitic neuropathy, amlyoidosis-associated neuropathy, MMN) [14]. The significance of NfL as a prognostic progression marker appears to be even greater than in differential diagnostics, although here too no reliable statement can be made for the individual patient on the basis of the NfL value.
While genetic diagnostics were still considered optional in the S1 guideline, which has been valid and available since 2021, the authors believe that this situation has changed as a result of developments over the last three years [5]. In view of therapeutic developments in this field, every ALS patient should undergo mandatory targeted genetic testing at least for the presence of a mutation in the Cu/Zn superoxide dismutase 1 gene(SOD 1) and young ALS patients under the age of 40 for the presence of a pathogenic mutation in the FUS gene [15,16]. Optionally, more extensive genetic diagnostics should also be carried out with the examination of other genes such as C9orf72, TARDP, TBK1 etc. [17]. The following sections on aetiology and genetics and, in particular, therapy will deal with this issue in more detail. Table 1 provides an overview of the diagnostic procedures described and the Gold Coast diagnostic criteria recommended as a guide in clinical practice .
With regard to further clinically relevant examinations for comprehensive differential and exclusion diagnostics depending on the initial clinical presentation (e.g. MRI imaging, ENT diagnostics, laboratory diagnostics) as well as relevant diagnostics in the course of prognosis assessment (e.g. pulmonary function test/diaphragmatic function test, swallowing diagnostics using FEES, psychometrics using ECAS), we would like to explicitly refer to the very detailed and clear presentation of the S1 guideline [5].
Phenotypic spectrum and pattern of muscle paresis against the background of neuroanatomy and pathophysiology of ALS
The scientific findings of the last two decades, in particular neuroanatomical and neuropathological studies, have fundamentally changed the view of ALS. Today, ALS is no longer seen as a purely motor system degeneration, but as a multi-system degeneration [18,19].
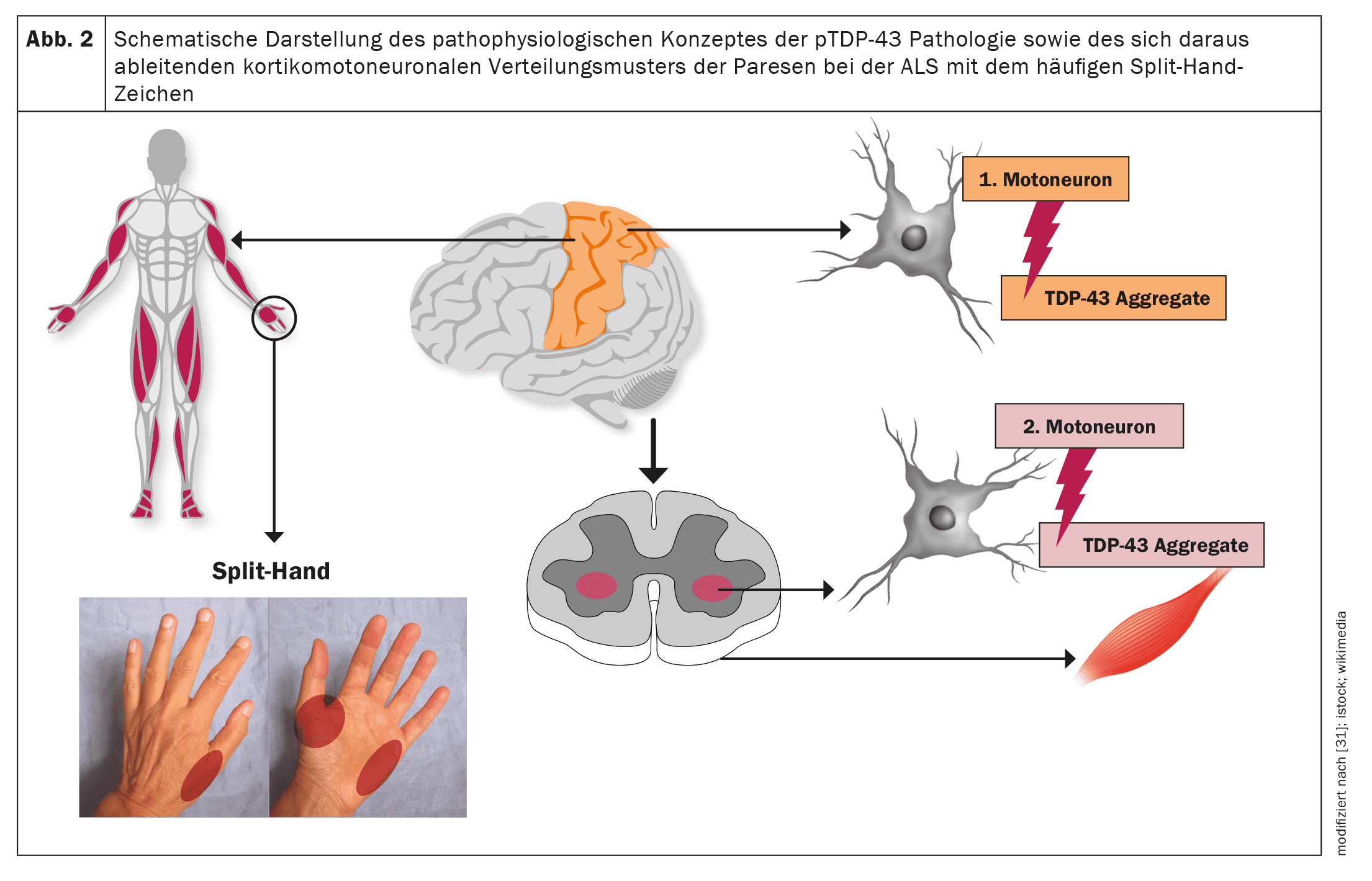
ALS, like other neurodegenerative diseases such as Alzheimer’s or Parkinson’s disease, is a proteinopathy, i.e. pathological protein aggregates in the motor nerve cells as a neuropathological correlate lead to dysfunction of the affected motor nerve cells and ultimately to apoptosis and thus the loss of motor nerve cells. In the case of ALS, this involves TDP-43 protein aggregates in more than 95% of cases [20]. Only in the case of underlying pathological SOD1 or FUS variantsdoes TDP-43-independent neurodegeneration occur [21]. Neuropathological studies have established a stepwise, staged, cerebral propagation of TDP-43 pathology in ALS, comparable to α-synuclein pathology in Parkinson’s disease and tau pathology in Alzheimer’s disease [22]. These findings as well as the corticomotoneuronal pattern of paresis with the split hand sign (asymmetric atrophy of the C8/T1 or ulnar-supplied hand muscles) as a very common clinical sign in ALS imply an origin of the neuropathological changes in the area of the primary motor cortex and a gradual spread of the TDP-43 pathology from there via axonal transport to the second motor neurons, i.e. the motor cranial nerve nuclei in the brain stem and the anterior horn cells of the spinal cord [23–26].
A prion-like propagation of the neuropathological changes appears to occur, which would explain the focal onset of the motor manifestation as well as a gradual spread to neighboring myotomes and body regions [27,28]. Accordingly, depending on the initial manifestation of the neuropathology, the phenotypic spectrum of ALS – with regard to motor symptoms alone – is not uniform, but highly variable [29]. Thus, topographically distinct motor phenotypes alone can occur. In addition, the phenotype is influenced by the usually different speed of degeneration of the first and second motor neurons and the associated clinical symptoms. Without a precise understanding of the molecular mechanisms to date, the ratio of insoluble TDP-43 protein aggregates, which can no longer be transported axonally and thus accumulate locally, to soluble TDP-43 oligomers as precursors of the TDP-43 aggregates appears to be decisive here. Obviously, these soluble precursors are then transported via axonal transport from the first motor neurons to the second motor neurons, where they are obligatory deposited as protein aggregates and lead to the death of these cells [23].
To date, there is no standardized classification of clinical phenotypes that would take into account the ratio or focus of damage to the first and second motor neuron, the clinical pattern or focus of atrophic paresis and its spread.
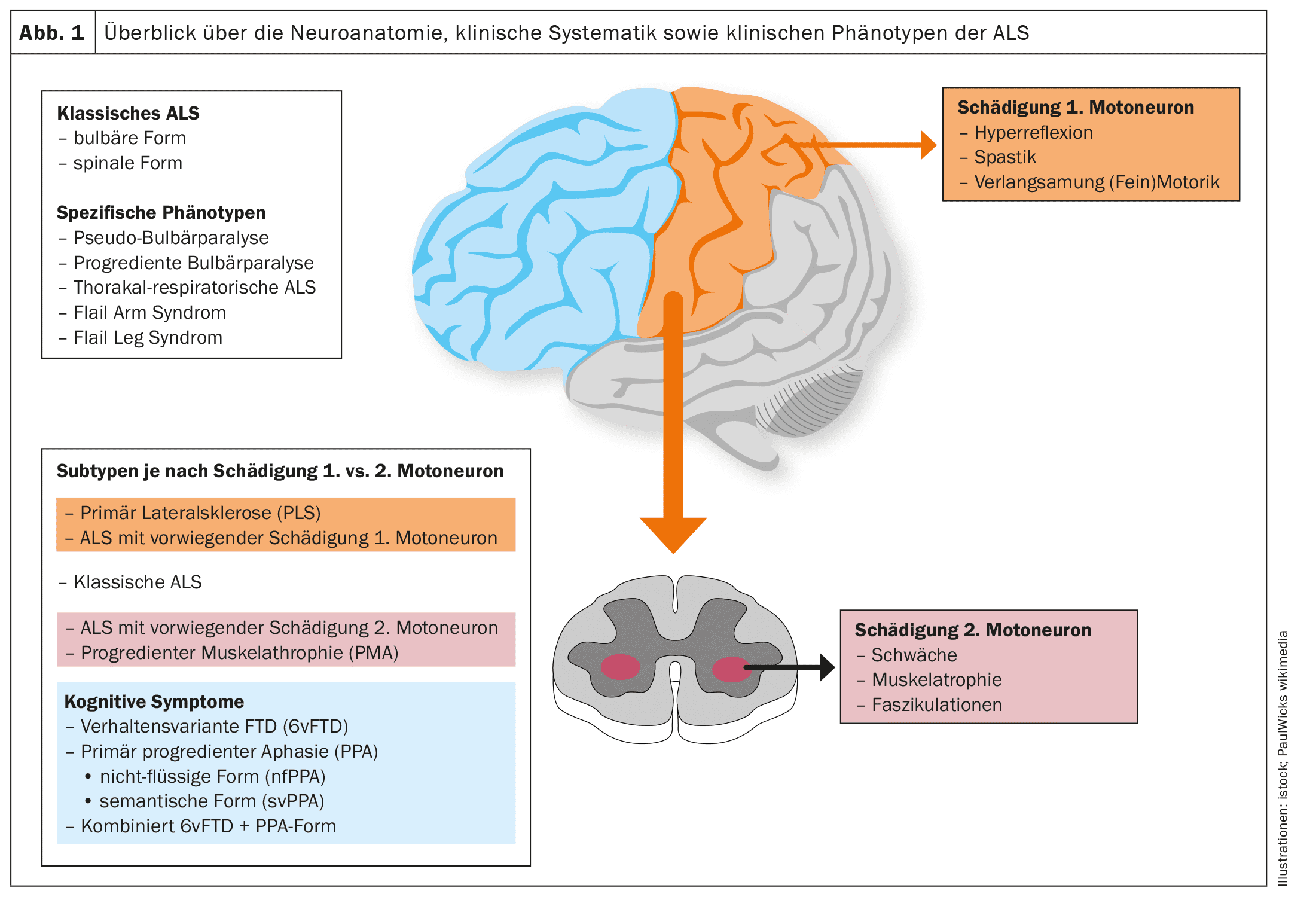
Figures 1 and 2 provide a schematic overview of the neuroanatomy and pathophysiology of the disease as well as the clinical phenotypes and their classification.
Etiology and genetics
The etiology of ALS is still not sufficiently well understood. In the sporadic form of the disease, which accounts for the vast majority of cases, a multifactorial genesis is assumed with the interaction of various unfavorable external environmental factors, epigenetic metabolic factors and a possible additional genetic susceptibility. Changes in nuclear-cytosolic protein transport, RNA metabolism, mitochondrial oxidative cell function, glutamatergic excitability, axonal protein transport and cellular autophagy are discussed as the underlying metabolic intracellular mechanisms, while functional disorders of atrocytes and oligodendrocytes as well as inflammatory processes are discussed as additive extracellular mechanisms [31]. Significant insights into the possible etiologic mechanisms of sporadic ALS have been gained primarily through genetic forms with distinct pathophysiologic processes.
In 1993, pathogenic mutations in Cu/Zn superoxide dismutase 1 (SOD1) were described as a genetic cause of ALS for the first time [32]. A large number of genetic mutations are now known to be the underlying etiology of ALS. In the case of apparently sporadic ALS without a conspicuous family history in Germany, a monogenetic cause can be detected in slightly more than 10% of cases [17]. The most common genetic causes in Germany are pathological C9orf72 repeat expansions and mutations in the SOD1, TARDP and FUS genes [17].
Genetic diagnosis at the time of diagnosis should therefore be offered to every ALS patient, as this can have immediate therapeutic consequences, which we will discuss in detail in the following section.
Therapy and prognosis
Riluzole has been approved in Germany since 1996 as the only pharmacological substance with a proven positive effect on the progression of ALS. A reduction in glutamate release and thus a reduction in excitotoxicity is postulated as the decisive mechanism of action. This hypothesis has been given additional substance by evidence of primary degeneration of glutamatergic corticofugal pathways [18]. A daily riluzole dose of 100 mg has the best efficacy/side-effect profile. Retrospective analyses from a total of ten clinical ALS registries provide evidence of a median extension of survival time by up to 19 months with riluzole, and there are also clear indications that this substance is also effective in later stages of the disease [33–35]. Riluzole is generally well tolerated; known side effects include an increase in transaminases, which must therefore be monitored regularly, and gastrointestinal complaints. Alternative dosage forms such as juice or orodispersible tablets are now also available for ALS patients with dysphagia.
In addition to pharmacological therapy with riluzole, the prevention of a catabolic metabolic state with consecutive weight loss is considered to have an additional prognostic benefit. The higher the body mass index (BMI), the more favorable the prognosis [36].
The current recommendations for ALS patients are therefore to maintain a stable weight and avoid weight loss. In this context, the insertion of a PEG tube also plays an important role in progressive dysphagia, with a prolonged survival time as a result of this measure [37]. Whether and to what extent specific anti-catabolic therapeutic approaches such as targeted high-fat, high-calorie nutritional interventions or ketogenic nutritional interventions can improve the prognosis is currently being intensively investigated in studies. In this context, the Germany-wide LIPCAL-ALS 2 study as an Investigator Initiated Trial (IIT) by colleagues from Ulm, which is scheduled to start in 2024 to 2025, is of great importance.
Non-invasive (NIV) and invasive ventilation by means of a tracheostoma is another important measure for prolonging survival in ALS with ventilatory insufficiency [38,39]. This is also plausible, as alveolar hypoventilation with consecutive hypercapnia due to diaphragmatic involvement is typically a significant factor in the death of ALS patients after three to five years of disease progression.
The development of antisense oligonucleotides (ASO) for specific genetically induced forms of ALS can be seen as a milestone. In particular, the intrathecally administered ASO Tofersen should be mentioned here, which binds the SOD1 mRNAin patients with pathogenic SOD1 variants(around 1-2% of all ALS cases) and thus prevents the cytotoxic expression of the SOD1 protein. It has been shown that the mechanistic effect of a significant reduction in SOD1 expressionby approx. 30% in humans occurs very rapidly within days of starting therapy with Tofersen, followed by a significant drop in NfL in CSF and serum, before finally a slowdown in the decline in the ALSFRS-R score [16,40]. After a longer-term observation of twelve months, further positive signals with clinical relevance such as effects on muscle function and weight stabilization were also observed.
In the USA, Tofersen was approved by the FDA in April 2023 solely on the basis of the convincing biomarker data with a significant drop in NfL values. In Germany, Tofersen was made available as part of an open access program. Initial real-world application data from the German motor neuron network impressively confirmed Tofersen’s study data with even better data in terms of clinical progression parameters [41]. In this context, it was only logical that the EMA decided to approve Tofersen in February 2024.
In addition to the Tofersen ASO for SOD1-associatedALS, ASOs are currently being developed or have already been investigated in studies, particularly against C9orf72 and FUS [15,42]. In particular, the ASO Jacifusen (ION363) should be mentioned here in the detection of a pathogenic FUS variant as the cause of juvenile ALS, which is currently being tested in a multicenter, multinational study with two study sites (Rostock and Ulm) in Germany.
The revised ALS-Functional Rating Scale (ALSFRS-R) is a proven and well-established score for evaluating the motor functions of all four body regions and is not only an important endpoint for clinical studies, but has also proven to be an easily feasible follow-up parameter in clinical practice [43]. The ALSFRS-SE (SE: “self-explanatory”), which was only recently agreed within the German MND network in German with corresponding concrete and exemplary explanations for the individual items and functional impairments, is expected to have an additional advantage in terms of practical handling and, in particular, diagnostic accuracy [44].
Conclusion for practice
In addition to standard care for all forms of ALS (Riluzole, prevention of catabolic metabolic state including timely insertion of a PEG tube, early non-invasive and, if necessary, invasive ventilation in the event of ventilatory insufficiency), there has been significant progress, particularly for monogenetic forms of ALS, thanks to the development of specific gene-based treatment methods, currently ASO in particular. Tofersen should be mentioned here as a very effective, specific therapy for SOD1-associatedALS, which is now also available in Germany.
Basic genetic diagnostics, at least of the most common genes (SOD1, C9orf72, FUS, TARDP), are therefore recommended for all ALS patients at the time of diagnosis. The extent to which targeted, anti-catabolic, high-calorie interventions can favorably influence the course of sporadic ALS will hopefully be shown by upcoming studies in this area.
Take-Home-Messages
- Standard care for all forms of ALS includes riluzole 100 mg/d, prevention of a catabolic metabolic state, including timely placement of a PEG tube, and early non-invasive ventilation in the event of respiratory insufficiency. timely placement of a PEG tube, as well as early non-invasive and, if necessary, invasive ventilation for ventilatory insufficiency.
- In addition, significant progress has been made, particularly for the monogenetic forms of ALS. The basis for this is the development of specific gene-based therapy methods, such as antisense oligonucleotides (ASO) in particular. Tofersen should be mentioned here as a very effective, specific therapy for SOD1-associatedALS.
- A basic genetic diagnosis, at least of the most common genes (SOD1, C9orf72, FUS, TARDP) is therefore recommended for all ALS patients at the time of diagnosis.
Literature:
- Duyckaerts C, Maisonobe T, Hauw JJ, Seilhean D: Charcot identifies and illustrates amyotrophic lateral sclerosis. Free Neuropathol. 2. doi:10.17879/freeneuropathology-2021-3323.
- Uenal H, Rosenbohm A, Kufeldt J, et al: Incidence and Geographical Variation of Amyotrophic Lateral Sclerosis (ALS) in Southern Germany – Completeness of the ALS Registry Swabia. PLoS ONE. 2014; 9(4). doi:10.1371/journal.pone.0093932.
- Jun KY, Park J, Oh KW, et al: Epidemiology of ALS in Korea using nationwide big data. J Neurol Neurosurg Psychiatry. 2019;90(4): 395-403. doi:10.1136/jnnp-2018-318974.
- Marin B, Boumédiene F, Logroscino G, et al: Variation in worldwide incidence of amyotrophic lateral sclerosis: a meta-analysis. Int J Epidemiol 2017; 46(1): 57-74. doi:10.1093/ije/dyw061.
- Petri SA-O GJ Ludolph AC. “Motor neuron diseases” of the German Society of Neurology (DGN). (2524-2348; Electronic).
- Finsel J, Uttner I, Vázquez Medrano CR, et al: Cognition in the course of ALS-a meta-analysis. Amyotroph Lateral Scler Front Degener. 2023; 24(1-2): 2-13. doi:10.1080/21678421.2022.2101379.
- Iazzolino B, Pain D, Peotta L, et al: Validation of the revised classification of cognitive and behavioral impairment in ALS. J Neurol Neurosurg Psychiatry 2019; 90(7): 734-739. doi:10.1136/jnnp-2018-319696.
- Oprisan AL, Popescu BO: Dysautonomia in Amyotrophic Lateral Sclerosis. Int J Mol Sci 2023; 24(19). doi:10.3390/ijms241914927.
- Chiò A, Mora G, Lauria G. Pain in amyotrophic lateral sclerosis. Lancet Neurol 2017; 16(2): 144-157. doi:10.1016/S1474-4422(16)30358-1
- Shefner JM, Al-Chalabi A, Baker MR, et al: A proposal for new diagnostic criteria for ALS. Clin Neurophysiol 2020; 131(8): 1975-1978. doi:10.1016/j.clinph.2020.04.005.
- Koch JC, Petri S, Zeller D: Electrophysiologic Diagnosis of Suspected Amyotrophic Lateral Sclerosis – Consensus Recommendations of the German Network for Motor Neuron Diseases (MND-NET). Clin Neurophysiol 2024. 2024; 55: 82-88.
- Zoccolella S, Mastronardi A, Scarafino A, et al: Motor-evoked potentials in amyotrophic lateral sclerosis: potential implications in detecting subclinical UMN involvement in lower motor neuron phenotype. J Neurol 2020; 267(12): 3689-3695. doi:10.1007/s00415-020-10073-5.
- Shahim P, Norato G, Sinaii N, et al. Neurofilaments in Sporadic and Familial Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis. Genes. 2024;15(4). doi:10.3390/genes15040496.
- Behzadi A, Pujol-Calderón F, Tjust AE, et al: Neurofilaments can differentiate ALS subgroups and ALS from common diagnostic mimics. Sci Rep 2021; 11. doi:10.1038/s41598-021-01499-6.
- Korobeynikov VA, Lyashchenko AK, Blanco-Redondo B, et al: Antisense oligonucleotide silencing of FUS expression as a therapeutic approach in amyotrophic lateral sclerosis. Nat Med 2022; 28(1): 104-116. doi:10.1038/s41591-021-01615-z.
- Miller TM, Cudkowicz ME, Genge A, et al: Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N Engl J Med. Published online September 22, 2022. doi:10.1056/NEJMoa2204705.
- Ruf WP, Boros M, Freischmidt A, et al: Spectrum and frequency of genetic variants in sporadic amyotrophic lateral sclerosis. Brain Commun 2023; 5(3). doi:10.1093/braincomms/fcad152.
- Braak H, Brettschneider J, Ludolph AC, et al: Amyotrophic lateral sclerosis–a model of corticofugal axonal spread. Nat Rev Neurol 2013; 9(12): 708-714. doi:10.1038/nrneurol.2013.221.
- Brettschneider J, Del Tredici K, Toledo JB, et al: Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann Neurol 2013; 74(1): 20-38. doi:10.1002/ana.23937.
- Neumann M: Molecular Neuropathology of TDP-43 Proteinopathies. Int J Mol Sci 2009; 10(1): 232-246. doi:10.3390/ijms10010232.
- Saberi S, Stauffer JE, Schulte DJ, Ravits J: “Neuropathology of amyotrophic lateral sclerosis and its variants.” Neurol Clin 2015; 33(4): 855-876. doi:10.1016/j.ncl.2015.07.012.
- Braak H, Braak E: Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol Aging 1995; 16(3): 271-278-284.
- Braak H, Ludolph AC: Neumann M, Ravits J, Del Tredici K. Pathological TDP-43 changes in Betz cells differ from those in bulbar and spinal α-motoneurons in sporadic amyotrophic lateral sclerosis.
Acta Neuropathol (Berl) 2017; 133(1): 79-90.
doi:10.1007/s00401-016-1633-2. - Eisen A, Braak H, Del Tredici K, et al: Cortical influences drive amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 2017; 88(11): 917-924. doi:10.1136/jnnp-2017-315573.
- Ludolph AC, Emilian S, Dreyhaupt J, et al: Pattern of paresis in ALS is consistent with the physiology of the corticomotoneuronal projections to different muscle groups. J Neurol Neurosurg Psychiatry 2020; 91(9): 991-998. doi:10.1136/jnnp-2020-323331.
- Menon P, Kiernan MC, Vucic S: Cortical hyperexcitability precedes lower motor neuron dysfunction in ALS. Clin Neurophysiol 2015; 126(4): 803-809. doi:10.1016/j.clinph.2014.04.023.
- Prasad A, Bharathi V, Sivalingam V, et al: Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front Mol Neurosci 2019;12. doi:10.3389/fnmol.2019.00025.
- Gosset P, Camu W, Raoul C, Mezghrani A: Prionoids in amyotrophic lateral sclerosis. Brain Commun 2022;4(3). doi:10.1093/braincomms/fcac145.
- Hardiman O, Al-Chalabi A, Chio A, et al: Amyotrophic lateral sclerosis. Nat Rev Dis Primer 2017; 3:17071. doi:10.1038/nrdp.2017.71.
- Masrori P, Van Damme P: Amyotrophic lateral sclerosis: a clinical review. Eur J Neurol 2020; 27(10): 1918-1929. doi:10.1111/ene.14393.
- Eisen A, Vucic S, Mitsumoto H: History of ALS and the competing theories on pathogenesis: IFCN handbook chapter. Clin Neurophysiol Pract 2023; 9: 1-12. doi:10.1016/j.cnp.2023.11.004.
- Rosen DR, Siddique T, Patterson D, et al: Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993; 362(6415): 59-62. doi:10.1038/362059a0.
- Bensimon G, Lacomblez L, Meininger V, Group the AS: A Controlled Trial of Riluzole in Amyotrophic Lateral Sclerosis. http://dx.doi.org/10.1056/NEJM199403033300901.
- Hinchcliffe M, Smith A: Riluzole: real-world evidence supports significant extension of median survival times in patients with amyotrophic lateral sclerosis. Degener Neurol Neuromuscul Dis 2017;7: 61-70. doi:10.2147/DNND.S135748.
- Miller RG, Mitchell JD, Moore DH: Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND).
Cochrane Database Syst Rev 2012; 2012(3).
doi:10.1002/14651858.CD001447.pub3. - Dupuis L, Pradat PF, Ludolph AC, Loeffler JP: Energy metabolism in amyotrophic lateral sclerosis. Lancet Neurol 2011; 10(1): 75-82. doi:10.1016/S1474-4422(10)70224-6.
- Cui F, Sun L, Xiong J, et al: Therapeutic effects of percutaneous endoscopic gastrostomy on survival in patients with amyotrophic lateral sclerosis: A meta-analysis. PLoS ONE 2018; 13(2). doi:10.1371/journal.pone.0192243.
- Dorst J, Ludolph AC: Non-invasive ventilation in amyotrophic lateral sclerosis. Ther Adv Neurol Disord 2019; 12: 1756286419857040. doi:10.1177/1756286419857040.
- Radunovic A, Annane D, Rafiq MK, et al: Mechanical ventilation for amyotrophic lateral sclerosis/motor neuron disease. Cochrane Database Syst Rev 2017; 2017(10). doi:10.1002/14651858.CD004427.pub4.
- Miller T, Cudkowicz M, Shaw PJ, et al: Phase 1-2 Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N Engl J Med 2020; 383(2): 109-119. doi:10.1056/NEJMoa2003715.
- Wiesenfarth M, Dorst J, Brenner D, et al: Effects of tofersen treatment in patients with SOD1-ALS in a “real-world” setting – a 12-month multicenter cohort study from the German early access program. eClinicalMedicine. 2024; 69. doi:10.1016/j.eclinm.2024.102495.
- Meijboom KE, Brown RH: Approaches to Gene Modulation Therapy for ALS. Neurotherapeutics 2022;19(4): 1159-1179. doi:10.1007/s13311-022-01285-w.
- Cedarbaum JM, Stambler N, Malta E, et al: The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (Phase III). J Neurol Sci 1999; 169(1-2): 13-21. doi:10.1016/s0022-510x(99)00210-5.
- Maier A, Boentert M, Reilich P, et al: ALSFRS-R-SE: an adapted, annotated, and self-explanatory version of the revised amyotrophic lateral sclerosis functional rating scale. Neurol Res Pract 2022; 4. doi:10.1186/s42466-022-00224-6.
| First published in neuro aktuell 2024; 38(8): 30-35. |
HAUSARZT PRAXIS 2024; 19(11): 12–17









