As the most common hereditary clotting disorder, Von Willebrand syndrome affects about one in a thousand people. With new diagnostic possibilities and findings in the field of molecular genetics, the question of the clinical significance of genetic analysis is increasingly being raised. This was also a hotly debated topic at the 63rd Annual Meeting of the American Society of Hematology (ASH).
Genetic testing in von Willebrand syndrome (von Willebrand Disease, vWD) is like love, everyone talks about it, but no one understands it exactly – with these words Emmanuel Favaloro, a renowned Australian researcher at the Institutes of Clinical Pathology and Medical Research at Westmead Hospital in Sydney, his lecture on ASH Annual Meeting 2021. And indeed, there is some uncertainty regarding the best course of action. While genetic analysis in type 1 von Willebrand syndrome does not make sense in most cases, it is recommended in type 2 and 3 patients in the new guidelines published in 2021 [2].
The von Willebrand Syndrome: The basics
Von Willebrand syndrome was first described in 1926 as “pseudohemophilia” in a young woman who bled to death from it during one of her first periods. The discoverer – nomen est omen – is Dr. Erik von Willebrand. The autosomal inherited disease is defined by quantitative or qualitative abnormalities of von Willebrand factor (vWF), the symptomatic prevalence is about 1/1000. Although men and women are formally affected equally, women are diagnosed two to three times more often due to gynecologic complications. Most often, von Willebrand syndrome manifests as increased mucosal bleeding; in severe cases, musculoskeletal hemorrhage is also present.
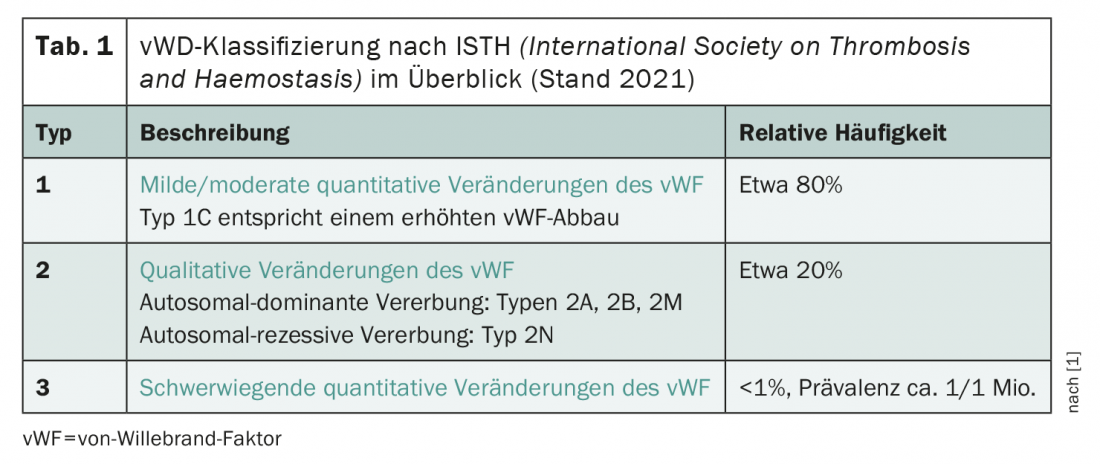
According to the recently revised classification of the International Society on Thrombosis and Haemostasis (ISTH), the clinical picture is roughly divided into three types (tab. 1) . In principle, a distinction is made between qualitative (type 2) and quantitative changes (types 1 and 3) of the vWF are distinguished. A new subtype 1C was added to the classification in 2021, which is defined by increased vWF degradation. The type 1 von Willebrand syndrome – that is, mild to moderate quantitative changes in vWF – is by far the most commonly diagnosed, followed by type 2. near-complete absence of vWF in von Willebrand syndrome type 3 is extremely rare, affecting about one in a million people.
In addition to symptomatic therapy using anti-fibrinolytics such as Cyklokapron and the pill in female sufferers, there are also treatment options that directly target the vWF. These consist of desmopressin (DDAVP), which promotes the release of vWF from endothelial cells, and recombinant or plasma-derived vWF. In principle, therapy is similar for all types of von Willebrand syndrome and depends on the severity. In more severe cases, the vWF usually needs to be replaced, as the effect of DDAVP decreases rapidly after only a short treatment period.
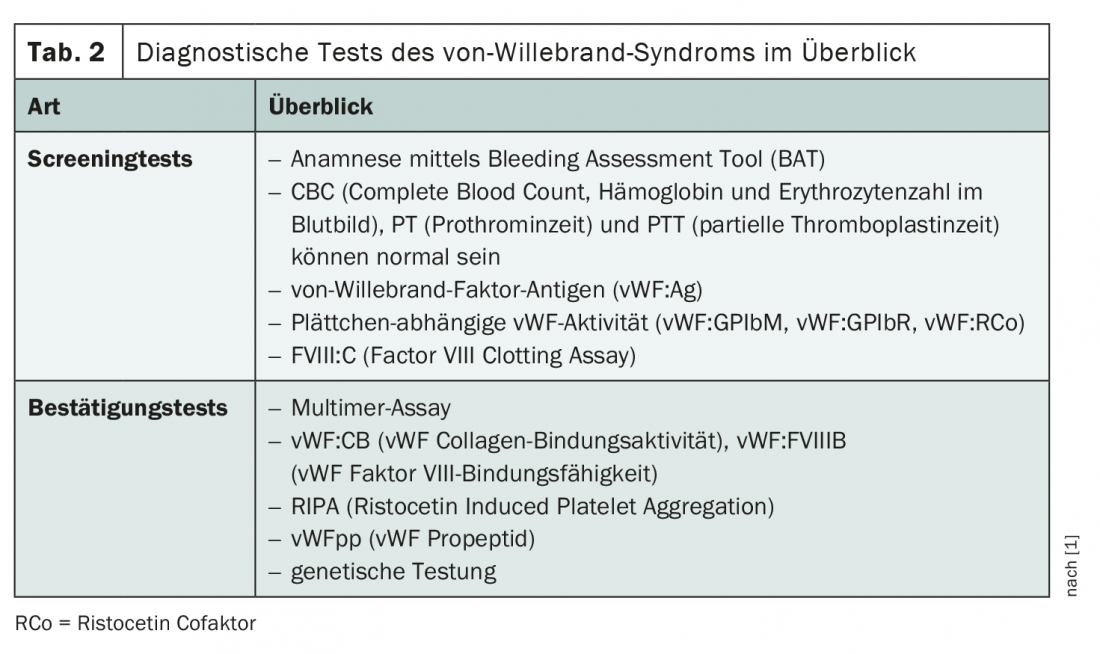
Various screening and confirmatory tests are used to establish the diagnosis (Tab. 2) . The medical history also plays a decisive role. Because blood counts, prothrombin time, and partial thromboplastin time may be normal, more specific screening procedures are needed. A panel of three diagnostic tools is usually performed to detect vWF antigens, platelet-dependent vWF activity, and factor VIII activity. If the suspicion of von Willebrand syndrome is confirmed during these investigations, further tests are used to confirm and identify the vWD subtype.
Genetic testing controversial
In the context of further testing, genetic analysis also plays a hitherto controversial role. Since therapy does not vary between subtypes and alternative, less expensive methods of classifying the disease are available, the question arises as to the usefulness of genetic characterization. Their availability varies, but is increasing. Not to be neglected are possible negative effects on the persons concerned, because the performance of genetic tests can, for example, lead to insurance-related problems and stigmatization. The bottom line is that genetic analysis for the most common vWD subtype, type 1, is usually not useful, according to experts at the ASH Annual Meeting – in part because a corresponding genetic variant can be detected in only about 65% of cases and has no impact on management.
Even if much has been done in terms of genetic characterization of the disease, which was first achieved in 1984, the database is still riddled with errors and gaps. For example, some so-called “variances of uncertain clinical significance” exist and certain genetic alterations could not yet be clearly assigned to their corresponding phenotypes. Nevertheless, one advantage of genetic testing is its central feasibility and thus good possibilities for standardization. The vWF gene is located on the short arm of chromosome 12.
When is genetic analysis useful?
Despite all reservations, there are some situations in which genetic testing in von Willebrand syndrome is already highly valued today. Especially for vWD subtypes 2B, 2N and 3, it can provide important, clinically relevant additional information – on the one hand for differentiation from other coagulation disorders and on the other hand for genetic counseling as well as assessment of therapy risk. Mutations can be detected in type 2 von Willebrand syndrome in about 90% of cases, and in type 3 in about 85% of cases. Regarding the optimal diagnostic approach including genetic analyses, new guidelines have been published in 2021 [2].
These recommend genetic testing for suspected vWD subtypes 2A, 2B as well as 2N [2]. The main issue here is the demarcation from the so-called “Platelet-type von Willebrand syndrome” (also known as “pseudotype”) and hemophilia A. This is because type 2B von Willebrand syndrome – typically characterized by increased affinity for platelets, abnormal multimer analysis and thrombocytopenia – strongly resembles the Platelet-type. However, while the causative mutation in the latter is located in the platelet gene, in type 2B von Willebrand syndrome it is located in the vWF gene. This has important implications for management: the “true” von Willebrand syndrome is treated by vWF replacement, the pseudotype by platelet administration. Platelet-type is inherited in an autosomal dominant manner. The cause is a gain-of-function mutation ofthe glycoprotein that binds vWF. This leads to excessive and unnecessary interaction between platelets and vWF with consecutive consumption of both components – and a clinical picture that often resembles the more common “true” von Willebrand syndrome type 2B is confused. It is estimated that about 15% of type 2B diagnoses in truth a platelet-type von Willebrand syndrome [3].
There is also an important differential diagnosis for type 2N (“Normandy”) von Willebrand syndrome: hemophilia A. In this subtype, factor VIII levels are classically lower than vWF levels – a constellation that in von Willebrand syndrome only occurs in type 2N occurs and is associated with hemophilia A can be confused. Thus, the new guidelines recommend that in cases of suspected type 2N vWD the targeted genetic testing and/or determination of vWF factor VIII binding ability [2]. Only by correctly identifying the underlying disease can adequate therapy – either by factor VIII or vWF substitution – be ensured.
Unlike type 2 von Willebrand syndrome, the diagnosis of type 3 usually clarified before genetic testing. Nevertheless, one may make sense in this rare, autosomal recessive form of the disease – although the localization of the triggering mutation varies more than in the genetically more clearly characterized types 2A, B and N. On the one hand, genetic analysis can play an important role in genetic counseling of affected individuals and their relatives, and on the other hand, it provides valuable information for risk assessment of therapy using exogenous vWF. Thus, large deletions predispose to allo-antibody formation during therapy. Also, patients with alterations in the vWF propeptide appear to be at greater risk of bleeding than those affected with mutations at other sites.
The bottom line is that the benefit of genetic testing in type 1 von Willebrand syndrome – and thus in more than 70% of all affected individuals – is currently questionable. In addition to the inadequately characterized genotype-phenotype correlation, the therapeutic consequence of such analyses is also lacking. For the types 2 and 3, on the other hand, genetic analysis can be quite useful and is already highly valued in differential diagnosis, risk assessment and genetic counseling.
Congress: 63rd ASH Annual Meeting
Source/Literature:
- Spotlight Session “Genetic Testing for von Willebrand Disease,” Paula James and Emmanuel Favaloro, 12/13/2021, 63rd ASH Annual Meeting, Atlanta, USA.
- James PD, et al: ASH ISTH NHF WFH 2021 guidelines on the diagnosis of von Willebrand disease. Blood Adv. 2021; 5(1): 280-300.
- Othman M: Platelet-type von Willebrand disease: a rare, often misdiagnosed and underdiagnosed bleeding disorder. Semin Thromb Hemost. 2011; 37(5): 464-469.
InFo ONCOLOGY & HEMATOLOGY 2022; 10(1): 38-40.