Grade II gliomas tend to be underestimated in their malignancy in the early stages. There is no standard for treatment. In addition to wait-and-see, aggressive surgical tactics are becoming more common. However, no cure can be achieved with it either.
Low-malignant gliomas WHO grade II frequently infiltrate the cortex and therefore lead to epileptic seizures in 60-80% of cases, often as the initial manifestation [1,2]. The largest lobe of the brain, the frontal brain, is most commonly affected. Personality changes, drive and mood disorders, and also frontal ataxia occur. If the frontoprecentral to postcentral region is affected, there are persistent focal motor seizures with a tendency to generalization, but also spastic hemiparesis and sensory disturbances. When the temporal lobe is affected, partial-complex seizures and even speech disorders predominate. Disorders of the visual system are rare, the most likely cause being homonymous hemianopsia when the tractus opticus is affected. If the brainstem is affected, there are complex neurological disturbances of the long pathways and cranial nerve function, including bulbar paralysis with dysphagia and aspiration. If thalamus and basal ganglia are affected, extrapyramidal disorders or Fluctuations in vigilance to dementia come to the fore. Often, however, personality is well preserved for a long time, even in very large processes, because the tumor cells diffusely grow through the healthy brain tissue without destroying it. The slow displacement of functional areas by tumor tissue allows the threatened function to shift to neighboring areas or to the opposite side (plasticity), which can be seen on functional MRI.
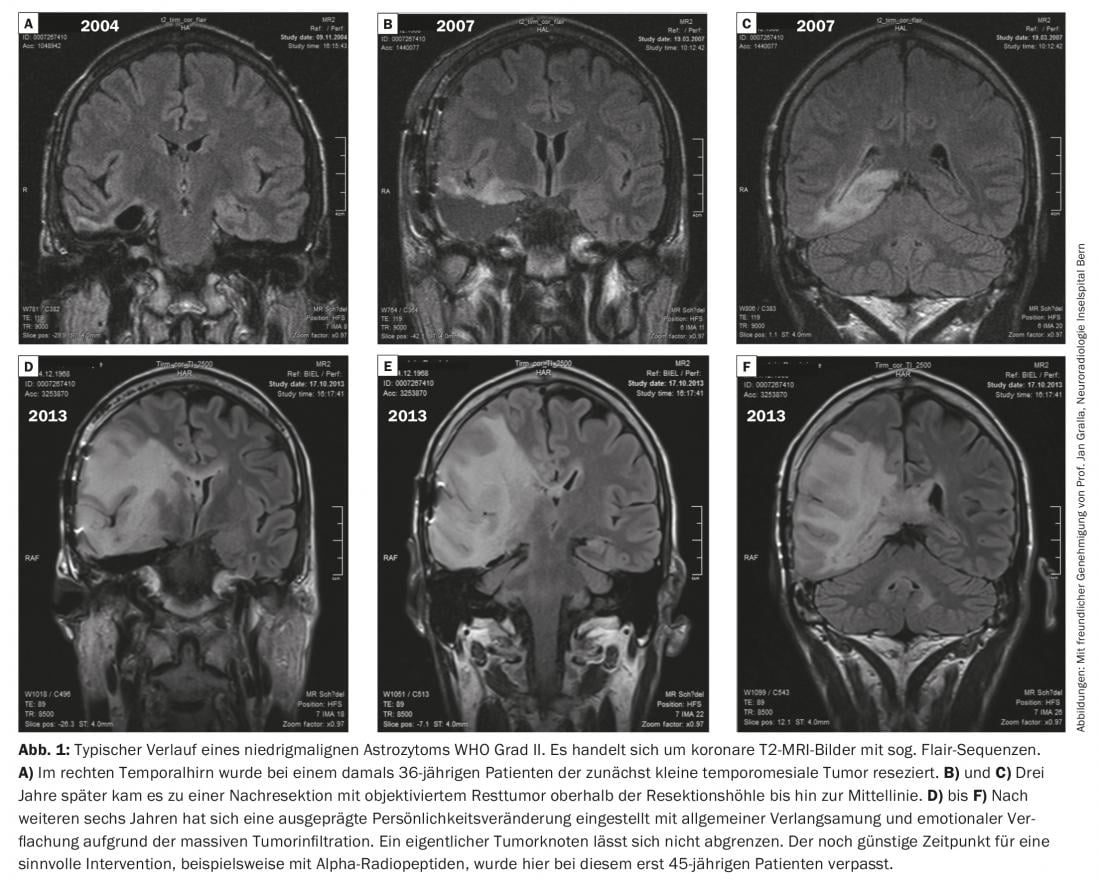
Figure 1 shows a typical course of a low-malignant astrocytoma WHO grade II.
Gliomas: Manifestation of spontaneous mutation rate?
Rarely (<5%), brain tumors occur syndromically in familial cancers, e.g., Turcot syndrome with “mismatch repair” gene defect or Li-Fraumeni syndrome with p53 mutation [3–5]. Even rarer are the benign giant cell astrocytomas manifesting in childhood and sporadically in adults as a consequence of a congenital mutation of the tuberous sclerosis genes TSC1 and TSC2, which co-regulate the mTOR complex of energy metabolism. The most common genetic defect associated with neurological disease is the mutation of the neurofibromatosis type 1 gene, with a frequency of 1:3000. These are intragenic microdeletions, which in half of the cases are not inherited but occur spontaneously. The NF1 gene locus on chromosome 17q11.2 is relatively unstable. The Nf1 phenotype includes the benign optic gliomas. Nf1 deletions have been detected in glioblastoma subtypes and contribute to glioma genesis in combination with other mutations. In grade II astrocytomas and in the prognostically better secondary glioblastoma, mutations of the isocitrate dehydrogenase genes IDH1 and IDH2 are frequently found. Like the 1p-19q co-deletion in oligodendroglioma, the IDH mutation marks a different biologic type of origin with better prognosis.
The spontaneous mutation rate is 1:100,000 per cell division. An organism undergoes about1014 cell divisions until full differentiation. During life, innumerable reading errors occur in every cell during cell division – also in the stem cell reservoir – which are mostly immediately corrected by cell-intrinsic repair mechanisms. However, there are always mutations that are not recognized or detected. not be repaired, which in rare cases contributes to tumorigenesis. Gliomas belong to the so-called “orphan diseases”, to the very rare diseases, with an incidence of less than five cases per 10,000 persons per year.
Molecular genetic classification
Newly developed genetic tests that scan the entire genome allow fairly accurate prognosis of all glioma types and correctly assign histologically unclear cases [6]. This involves scanning the entire genome for pathological methylation patterns. The expression of many genes that control tumor cell proliferation and promote apoptosis is regulated by so-called CpG islands in the promoter region of the genes, which can be switched off by methylation, e.g. in organ differentiation, but also in cancer cells. Another type of gene inactivation represents the long-range deletion of chromosomal arms, converting a heterozygous section into a monozygous section, often with reduplication. One can now create a so-called allelotype over all chromosomes to detect the loss of heterozygosity. For example, monosomy 1p and 19q are pathognomic for slow-growing oligodendrogliomas. Wild-type oligodendrogliomas with 1p/19q heterozygosity behave much more aggressively. In glioblastomas, chromosome 10 monosomy and chromosome 7 trisomy with EGFR amplification are typically found. The combined analysis of allelotype and methylome allows a very precise classification of all gliomas, especially in cases where histology raises questions. If so-called long-term survivors are observed in glioblastoma studies, the histological diagnosis should be verified with molecular testing, as the histological error rate in large-scale studies is up to 7% [7].
Delayed mass reduction at manifestation of symptoms
Because most grade II glioma patients are in good clinical condition at diagnosis, an observation phase is often initially chosen [1,2]. Many patients can thus lead a normal life with relatively few symptoms for a few years. With this strategy, intervention is only performed if the malignancy has progressed to a higher grade (grade III or IV), or if the tumor has become a malignant tumor (grade III or IV). if neurological symptoms manifest themselves up to signs of cerebral pressure (headache, vigilance disorders, nausea to vomitus). Mass reduction is then scheduled, followed by radiochemotherapy, or resigned.
Awake craniotomy and neuromonitoring
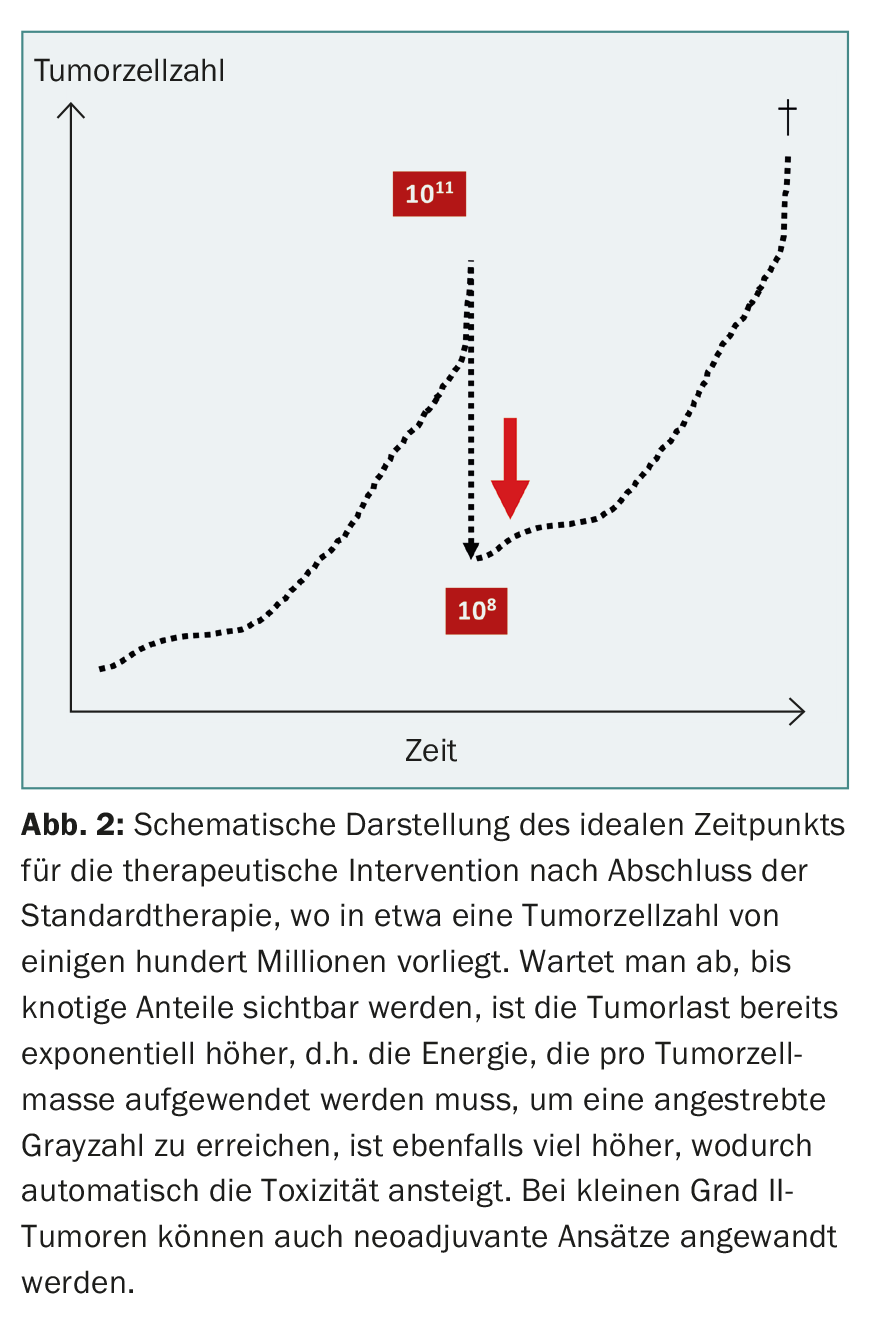
In the early phase of glioma surgery, attempts were also made in desperate cases to control the tumor by hemispherectomy, which was unsuccessful because of the diffuse infiltrative tendency of the tumor cells. In recent years, aggressive glioma surgery has experienced a renaissance, albeit limited to extensive mass reduction via awake craniotomy and neuromonitoring [8]. Cortical reorganization due to cerebral plasticity allows occasional resection of functionally important areas without additional deficits. Logically, this approach cannot lead to a cure either, because millions of infiltrative tumor cells remain undetected. This can be shown using a model calculation: A human body with 70 kg body weight consists of approx.1014 cells. Consequently, a tumor of 70 g contains approximately 1011 tumor cells. With a 99.9 percent resection of an infiltrative malignancy, about 100 million tumor cells still remain, which determine the further fate (Fig. 2) . Modern therapy must eliminate these residual cells or prevent them from developing further. can control.
Therapeutic outlook
Many grade II astrocytoma patients do not exceed the age of 50 resp. only with increasing neurological deficits and face the constant threat of a 50% chance of transformation to a higher malignancy. This actually argues for early intervention. Supramaximal resection does not result in cure but reduces the risk of malignant transformation. Initial experience with targeted single-cell irradiation by diffusible biomolecules that dock to specific tumor cell receptors and carry a very short-range, high-energy isotope as an effector has repeatedly resulted in very prolonged tumor control, well beyond median survival, without significant toxicity [9,10]. Other drugs will be added in the future that block the disrupted biological regulatory circuits and thus improve the prognosis [5].
Take-Home Messages
- Approximately 10-15% of all malignant gliomas occur primarily as low-malignant grade II astrocytomas or, less commonly, as oligodendrogliomas. Median survival is seven to ten years for grade II astrocytomas and ten to 15 years for grade II oligodendrogliomas.
- Considering the abysmal prognosis of the more common glioblastomas, grade II gliomas tend to be underestimated in their malignancy in the early stages.
- Brain tumors are probably a consequence of the spontaneous mutation rate of cellular processes, in a sense the flip side of recombination.
- The new WHO classification of brain tumors based on methylation pattern and allelotype allows reliable prediction of prognosis.
- There is no standard of care for the treatment of grade II gliomas. In addition to the wait-and-see approach, aggressive surgical tactics are increasingly being adopted. However, because of tumor cell infiltration of healthy brain tissue, no cure can be achieved. There is preliminary evidence that targeted single-cell radiotherapy with diffusible biomolecules may substantially improve prognosis.
Literature:
- Merlo A, de Tribolet N: Tumors of the brain and spinal cord. In: Steck A, Hess CH (eds.): Neurological Pathophysiology. Bern: Verlag Hans Huber 2003.
- Schneider T, et al: Gliomas in adults. Dtsch Arztebl Int 2010; 107(45): 799-807.
- Merlo A, Rochlitz C, Scott R: Survival of patients with Turcot’s syndrome and glioblastoma [letter]. N Engl J Med 1996; 334(11): 736-737.
- Merlo A, Bettler B: Glioblastomas on the move. Science STKE 2004; 2004(229): pe18.
- Lino M, Merlo A: Translating Biology into Clinic: the case of glioblastoma. Curr Opin Cell Biol 2009; 21(2): 311-316.
- Louis DN, et al: The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 2016; 131(6): 803-820.
- Linz U: Commentary on Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III trial: 5-year analysis of the EORTC-NCIC trial (Lancet Oncol. 2009;10:459-466). Cancer 2010; 116(8): 1844-1846.
- Duffau H: The Rationale to Perform Early Resection in Incidental Diffuse Low-Grade Glioma: Toward a “Preventive Surgical Neurooncology”. World Neurosurgery 2013; 80(5): e115-e117.
- Cordier D, et al: Targeted alpha-radionuclide therapy of functionally critically located gliomas with 213Bi-DOTAGA-substance P: a pilot trial. Eur J Nucl Med Mol Imaging 2010; 37(7): 1335-1344.
- Cordier D, et al: Targeted Radiolabeled Compounds in Glioma Therapy. Semin Nucl Med 2016; 46(3): 243-249.
InFo ONCOLOGY & HEMATOLOGY 2017; 5(6): 7-10.