Pulmonary hypertension is divided into 5 distinct groups, with group 1 reflecting pulmonary arterial hypertension (PAH). The PH-specific drug therapies approved to date interfere with three different signaling pathways (NO signaling pathway, prostacyclin signaling pathway, and endothelin receptor signaling pathway). Much has happened since the first PAH-specific drug (bosentan) was approved in Europe about 20 years ago. Numerous other agents have been added and have improved the prognosis of PAH patients.
Pulmonary hypertension is divided into 5 distinct groups, with group 1 reflecting pulmonary arterial hypertension (PAH). The PH-specific drug therapies approved to date interfere with three different signaling pathways (NO signaling pathway, prostacyclin signaling pathway, and endothelin receptor signaling pathway). Much has happened since the first PAH-specific drug (bosentan) was approved in Europe about 20 years ago. Numerous other agents have been added and have improved the prognosis of PAH patients.
While idiopathic PAH (IPAH) was long primarily diagnosed in young patients without clinically relevant comorbidities, a demographic shift has become apparent in recent years. Currently, an increasing number of elderly patients with pulmonary and/or cardiac comorbidities are diagnosed with severe precapillary pulmonary hypertension and are classified as IPAH patients based on the current classification. In this regard, various database analyses [1,2] have revealed that IPAH patients have very different disease characteristics and different clusters or phenotypes exist. It should be emphasized that diffusion capacity (DLCO), among other factors, is a crucial discriminatory and predictive factor [3–5].
This article reproduces the lecture “PAH: what is there beyond the new guideline” given at the 63rd German Congress of Pneumologists in Düsseldorf on March 30, 2023. The presentation demonstrated study data from patients diagnosed with IPAH who had mild pulmonary skeletal change on CT thorax and/or severe alveolocapillary diffusion defect of unclear etiology.
Comorbidities as a prognostic factor – current study situation.
Already in 2016, during the Cologne Consensus Conference, it was discussed among German-speaking PH experts that the IPAH patient population included in prospective, multicenter, and placebo-controlled therapy trials is often not comparable to the patients included in various national PH registries and also dominate in clinical practice. It was already noticeable at that time that the latter were older, had more comorbidities, and were phenotypically similar to patients with left heart and/or lung disease because of their risk factors, but were classified as IPAH according to the definition criteria that are also currently valid. At that time, the terms “typical” and “atypical” IPAH patients were introduced to reflect this development. “Typical IPAH” was defined hemodynamically at that time, after exclusion of all known causes of PH, by a mean pulmonary arterial pressure (mPAP) ≥25 mmHg and a pulmonary arterial occlusion pressure (PAWP) ≤15 mmHg. Furthermore, similar to the AMBITION study (ambrisentan plus tadalafil versus ambrisentan or tadalafil monotherapy), only a maximum of two risk factors for left ventricular diastolic dysfunction (Heart Failure with preserved left-ventricular ejection fraction, HFpEF: arterial hypertension, coronary artery disease, diabetes mellitus, and obesity with BMI >30 kg/m2) must be present and the diffusion capacity (DLCO) must be at least 45% of the nominal value. [6,7]. In the group of “atypical IPAH,” which did not differ in hemodynamic profile from “classic IPAH,” patients were predominantly older (mostly >65 years) and had the risk profile or concomitant diseases of patients with left heart or lung disease. At that time, they were referred to as “IPAH patients with cardiac phenotype” and “IPAH patients with pulmonary phenotype,” respectively.
Patients with a pulmonary phenotype were characterized by (near) normal body thysmography and CT chest findings, often severe hypoxemia, and marked diffusion capacity reduction (DLCO <45% of the target value) ( Table 1) . With regard to targeted PH therapy, monotherapy was initially recommended in the “atypical” patients at that time, as the efficacy and tolerability of PAH-specific drugs in this population had not been investigated sufficiently to be able to make evidence-based recommendations. Based on the registry data, it was evident that these patients were predominantly treated with PDE5 inhibitors and also acted rather cautiously with regard to combination therapy during the course.

DLCO as a prognostic risk factor – current study situation.
Back in 2010, a study was published [4] describing an association between DLCO and survival in PAH patients. Here, three ranges for DLCO (DLCO >64%, DLCO 43-63%, DLCO <43%) were elaborated that discriminated well the risk of dying. In this study, decreased DLCO was multivariately associated with patient age, presence of collagenosis, decreased functional exercise capacity, oxygen demand, and reduced lung volumes and CT morphologic changes. These correlations were independent of cardio-pulmonary hemodynamics. Patients who had the lowest DLCO range (<43%) had a 2.7-fold increased risk of death.
In 2013, Trip et al. [8] that IPAH patients with a DLCO <45% have a significantly worse prognosis compared with IPAH patients with a higher DLCO age-corrected. Overall, approximately 75% of all IPAH patients had decreased DLCO (predominantly mild to moderate in severity). The analysis excluded patients with known causes of severe diffusion reduction, such as patent foramen ovale, collagenoses, PVOD (pulmonary veno-occlusive disease), severe left heart and lung disease. Mild and moderate CT changes did not exclude the diagnosis of IPAH in this study.
Trip et al. demonstrated that patients with IPAH + DLCO <were 45% older on average (67 years vs 49 years) and predominantly male, had clustered CHD, and were more frequent smokers. Pulmonary functional parameters (FEV1, FEV1/FVC, and TLC) tended to be lower, and CT morphologic changes (mild to moderate emphysema or fibrosis) were more common compared with patients with a DLCO of ≥45%. In contrast, pulmonary hemodynamics were comparable. Nevertheless, these patients had poorer functional exercise capacity (6-minute walk test).
Survival at 3 years (DLCO <45% vs DLCO ≥45%) was 54% and 86%, respectively, and at 5 years was 30% and 80%, respectively. The authors concluded from their results that the severe DLCO reduction was probably due to a PH subtype to be considered separately, which could be triggered by cigarette smoke inhalation.
In 2017, the Hanover-based research group led by Olsson et al. [5] that IPAH patients with completely normal lung function and unremarkable CT chest findings but with a significantly reduced DLCO (<45% of target) are prognostically comparable to CPFE (combined pulmonary fibrosis and emphysema) patients. Both groups included predominantly men with smoking histories who did not differ in their characteristics, except for pulmonary functional and CT morphologic changes.
All patients were treated with PDE5 inhibitors in this study, but both walking distance in the 6-minute walk test and partial pressure of oxygen (paO2) tended to worsen comparably with therapy in both groups. Mortality in both groups did not differ significantly at 1, 2, and 4 years (80%/76%/38% vs. 64%/42%/42%). The authors concluded that the cause of the severe DLCO reduction in their IPAH patients was probably due to “smoking-related pulmonary vaculopathy.”
As shown by data from the Sheffield PH Registry (ASPIRE; United Kingdom), IPAH patients with mild parenchymal changes on CT thorax fail to improve exercise capacity despite combined PAH-specific therapy. These patients also had a significantly worse prognosis than IPAH patients without CT thoracic abnormalities. In IPAH patients without CT changes but with a DLCO <45%, the age-adjusted prognosis in this registry study was also significantly worse than in IPAH patients with a DLCO ≥45% [1].
2020 IPAH patients in the European COMPERA (Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension) database were subjected to cluster analysis based on the variables age, sex (male vs. female), smoking status ([Ex]Smokers: yes vs. no), DLCO (<45% vs. ≥45% of set point) and the presence vs. absence of at least one risk factor for left ventricular diastolic dysfunction (obesity, arterial hypertension, coronary artery disease, and diabetes mellitus). The clusters found were analyzed in terms of their baseline characteristics, their response to PH-specific therapies (changes in 6-minute walk distance, functional class, and blood levels of biomarkers), and prognosis, among others. Three different clusters were found (Fig. 1; clusters 1-3) .
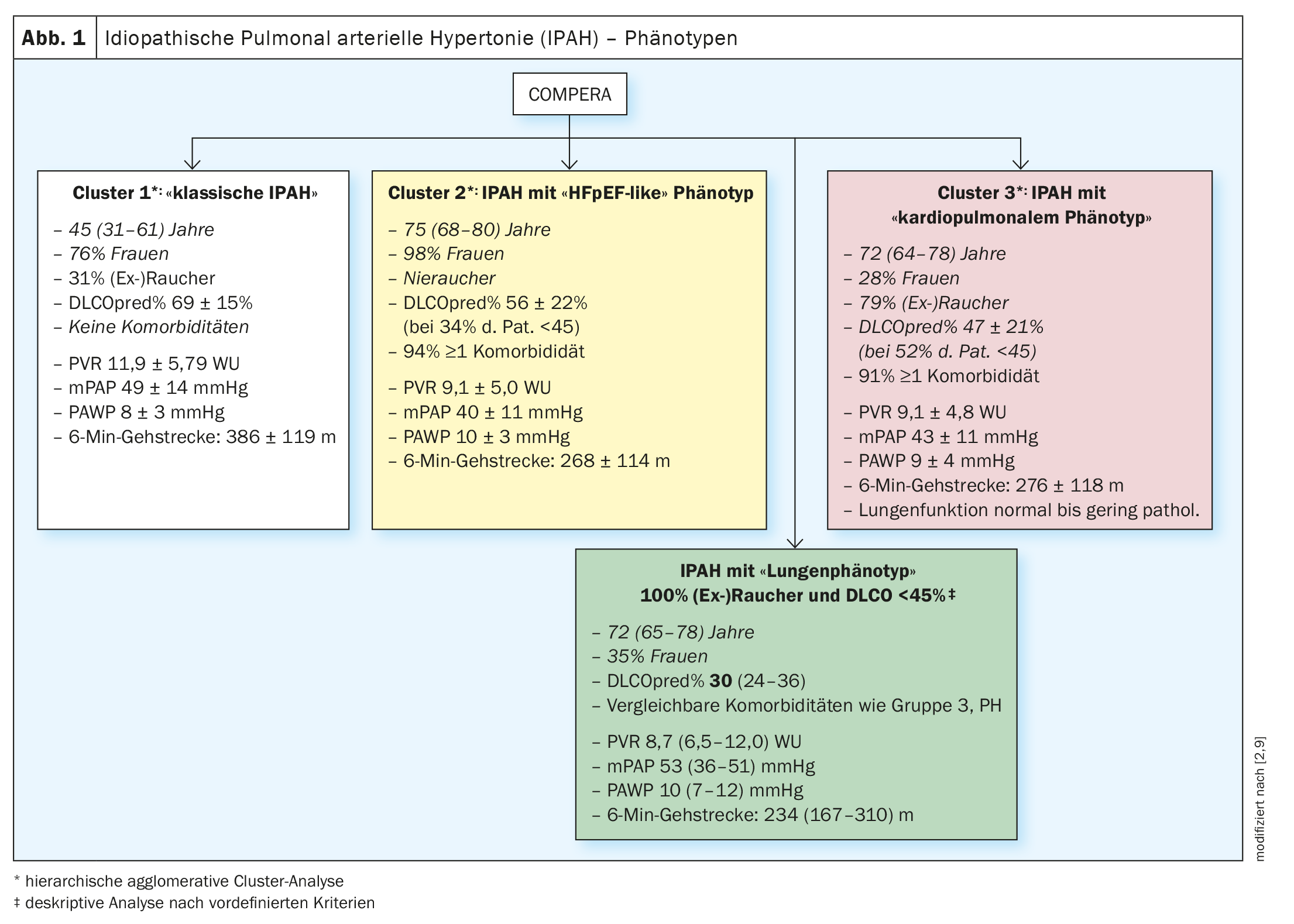
All three clusters were characterized by the presence of severe precapillary PH (PAWP averaged less than 11 mmHg in all 3 groups) and (largely) normal pulmonary function, with cluster 3 patients having the lowest values for pulmonary functional paramaters in comparison and the most pronounced hypoxemia. Adjusted for age, cluster 3 patients (IPAH patients with cardiopulmonary phenotype) had a significantly worse prognosis compared with cluster 1 patients (IPAH patients without comorbidities) [2].
While not all patients in cluster 3 in the COMPERA registry had a DLCO <45% (53% of cases) and positive (ex)smoking status (79% of cases) in this cluster analysis, in another PH registry study, a DLCO <45% and positive (ex)smoking status were necessary by definition for a diagnosis of IPAH with pulmonary phenotype [9]. For this study, the database of the COMPERA registry was again primarily analyzed and the ASPIRE registry served as an independent validation of the results [9].
In this study, it was shown that the group of IPAH patients with “pulmonary phenotype” resembled much more the patients of group 3-PH (PH associated with significant lung disease) in terms of treatment response and prognosis than the patients with so-called “classical” IPAH (IPAH without risk factors for HFpEF) [9].
Analysis of COMPERA data revealed, as in the previous analysis [2], that patients with “classic” IPAH (without risk factors for HFpEF) were predominantly younger (median 45 years) and mostly women (77%). Approximately 1/3 of these had a smoking history with a median of 14 pack years (py), but preserved lung function, and only mildly reduced DLCO (69% median). Hemodynamically, these patients were characterized by severe precapillary pulmonary hypertension (mPAP 48 mmHg, PVR 10.9 wood units (WU) and moderately reduced functional exercise capacity (6-min walk distance: 410 m) [9].
In comparison, IPAH patients with pulmonary phenotype were older (median 72 years) and more often male (65%). All patients with pulmonary phenotype were smokers by definition and had a median of 40 py. FVC and FEV1 were median in the lower normal range or slightly reduced and significantly lower than in patients with classic IPAH. DLCO was significantly impaired at 30% median, and paO2was 56 mmHg median. Hemodynamics were less compromised (mPAP 43 mmHg, PVR 8.7 WU) compared with classic IPAH patients, but functional exercise capacity (6-minute walk distance) was significantly more impaired (234 m) (Fig. 1 – green box).
Patients in group 3-PH in this study were comparable in median age to those with pulmonary phenotype (71 years vs. 72 years median) and had a comparable gender distribution (63% vs. 65% men) [9]. 81% were (ex)smokers with a median of 40 py, whereas here there were pulmonary functional limitations of FEV1 and FVC and DLCO was most reduced with a median of 26%. In contrast, hemodynamics were comparatively less restricted (mPAP 39 mmHg and PVR 7.4 WU median), and functional exercise capacity (6-minute walk distance) was comparable to that of patients with pulmonary phenotype (238 m vs 234 median).
CT morphologic changes in terms of emphysema or interstitial lung disease in the ASPIRE patient group increased significantly (from 10% to approximately 60%) from the group of “classic” IPAH patients to group 3 PH patients [9].
All three PH groups showed significant differences in terms of treatment response and prognosis. In classic IPAH patients compared with pulmonary phenotype IPAH patients and compared with group 3 PH patients, the 3-year survival probabilities were 90% vs. 49% vs. 43% and the 5-year survival probabilities were 84% vs. 31% vs. 26%. The authors suggested that the cause of right heart failure in the patients characterized as having IPAH with a pulmonary phenotype was cigarette smoke-induced pulmonary hypertension and concluded that these patients should probably be classified more as group 3 PH patients and that the place of PH-specific medications in these patients is unclear [9].
Summary
In summary, it suggests that (ex)smoking patients who meet current diagnostic criteria for IPAH but have a very severe DLCO reduction that cannot be explained otherwise and/or even only low-grade parenchymal abnormalities on CT thorax are likely to have a different PH group (most likely PH group 3) than patients with classic IPAH (PH group 1).
Presumably, the severe right heart failure in these patients is caused by pulmonary artery damage from cigarette smoke inhalation. The use of PH-specific medications is likely to be much less effective in patients with a most likely smoker-associated form of pulmonary hypertension than in classic IPAH patients.
Because these patients had been either excluded or underrepresented in previous pivotal trials of PH-specific drugs because of inclusion and exclusion criteria, prospective, randomized, placebo-controlled trials are urgently needed both to characterize these patients more precisely and to clarify treatment options and the place value of PH-specific drugs.
Take-Home Messages
- The current group of IPAH patients is more heterogeneous than previously thought.
- Patients who meet the criteria for IPAH but show evidence of smoker-associated PH should receive more scientific focus in the future for clarification of which PH group these patients belong to and what effective treatment options exist.
- Prospective, placebo-controlled, randomized treatment trials are urgently needed to clarify treatment options for presumed smoker-associated PH.
Literature:
- Lewis RA, Thompson AAR, Billings CG, et al: Mild parenchymal lung disease and/or low diffusion capacity impacts survival and treatment response in patients diagnosed with idiopathic pulmonary arterial hypertension. European Respiratory Journal; doi: 10.1183/13993003.00041-2020.
- Hoeper MM, Pausch C, Grünig E, et al: Idiopathic pulmonary arterial hypertension phenotypes determined by cluster analysis from the COMPERA registry. Journal of Heart and Lung Transplantation 2020; 39: 1435-1444; doi: 10.1016/j.healun.2020.09.011.
- Trip P, Nossent EJ, De Man FS, et al: Severely reduced diffusion capacity in idiopathic pulmonary arterial hypertension: patient characteristics and treatment responses. European Respiratory Journal; doi: 10.1183/09031936.00184412.
- Chandra S, Shah SJ, Thenappan T, et al: Carbon monoxide diffusing capacity and mortality in pulmonary arterial hypertension. Journal of Heart and Lung Transplantation 2010; 29: 181-187; doi: 10.1016/J.HEALUN.2009.07.005.
- Olsson KM, Fuge J, Meyer K, et al: More on idiopathic pulmonary arterial hypertension with a low diffusing capacity. European Respiratory Journal 2017; 50; doi: 10.1183/13993003.00354-2017.
- Hoeper MM, Apitz C, Grünig E, et al: Targeted therapy of pulmonary arterial hypertension. DMW 2016; S33-S41.
- Hoeper MM, Apitz C, Grünig E, et al: Targeted therapy of pulmonary arterial hypertension: updated recommendations from the Cologne Consensus Conference 2018. Int J Cardiol 2018; 272: 37-45; doi: 10.1016/j.ijcard.2018.08.082.
- Trip P, Nossent EJ, De Man FS, et al: Severely reduced diffusion capacity in idiopathic pulmonary arterial hypertension: patient characteristics and treatment responses. European Respiratory Journal 2013; 42: 1575-1585; doi: 10.1183/09031936.00184412.
- Hoeper MM, Dwivedi K, Pausch C, et al: Phenotyping of idiopathic pulmonary arterial hypertension: a registry analysis. Lancet Respir Med 2022; 10(10): 937-948; doi: 10.1016/S2213-2600(22)00097-2.
COIs:
- Halank: fees for lecturing and consulting activities from AstraZeneca, Janssen and MSD. Janssen travel expenses. All not in connection with the current review paper
- Heberling: honorarium for lecturing and consulting activities and travel expenses from Janssen-Cillag and MSD.
- Kolditz: none in connection with this work
- Koschel: none in connection with this work
InFo PNEUMOLOGIE & ALLERGOLOGIE 2023; 5(3): 6–11.