There are a variety of different mutations that can underlie hereditary retinal dystrophy. Thanks to the development of modern molecular genetic testing methods, the genetic cause can nowadays be identified in the majority of cases. An innovative gene therapy is available for patients with retinal dystrophy due to biallelic RPE65 mutations.
The umbrella term “hereditary retinal dystrophies” (IRD; inherited retinal diseases) encompasses a heterogeneous group of diseases with overlapping phenotypes characterized by progressive degeneration and dysfunction of the retina [1]. What these diseases have in common is that they affect the entire retina during the course of the disease and that vision deteriorates slowly and progressively over the course of a person’s life. IRD can be caused by mutations in >250 different genes [2]. Both the molecular pathogenesis and the clinical picture and course of the disease are significantly defined by the nature and type of the genetic defect [3]. Depending on the mutated gene, different developmental and functional disorders of the retina may occur, such as disturbances of retinal metabolism, cell structures or phototransduction. The most common form of IRD is retinitis pigmentosa, and there are also Leber’s congenital amaurosis and retinal dystrophy with early onset (EORD; early-onset retinal dystrophy) [2].
Early diagnosis is crucial
In retinitis pigmentosa, degeneration of the rods occurs in the early stages of the disease, resulting in night blindness. Characteristic symptoms of Leber’s congenital amaurosis include a dramatic reduction in visual acuity and visual field loss. Early correct diagnosis of the disease is very important as well as a detailed morphological and functional description of the current state of the retina depending on the underlying mutations in the RPE65 gene. This is because the rapid and severe progression of the disease makes early intervention necessary as long as the cells of the retinal pigment epithelium as target cells and also the photoreceptors are still present. Clinical description of the phenotype is possible in the context of ophthalmic functional diagnostics and especially noninvasive imaging diagnostics (e.g., optical coherence tomography, fundus or near-infrared autofluorescence) [5,6]. To identify the genetic cause of hereditary retinal dystrophies, the use of molecular genetic techniques such as next-generation sequencing is essential.
Autofluorescence imaging provides important findings
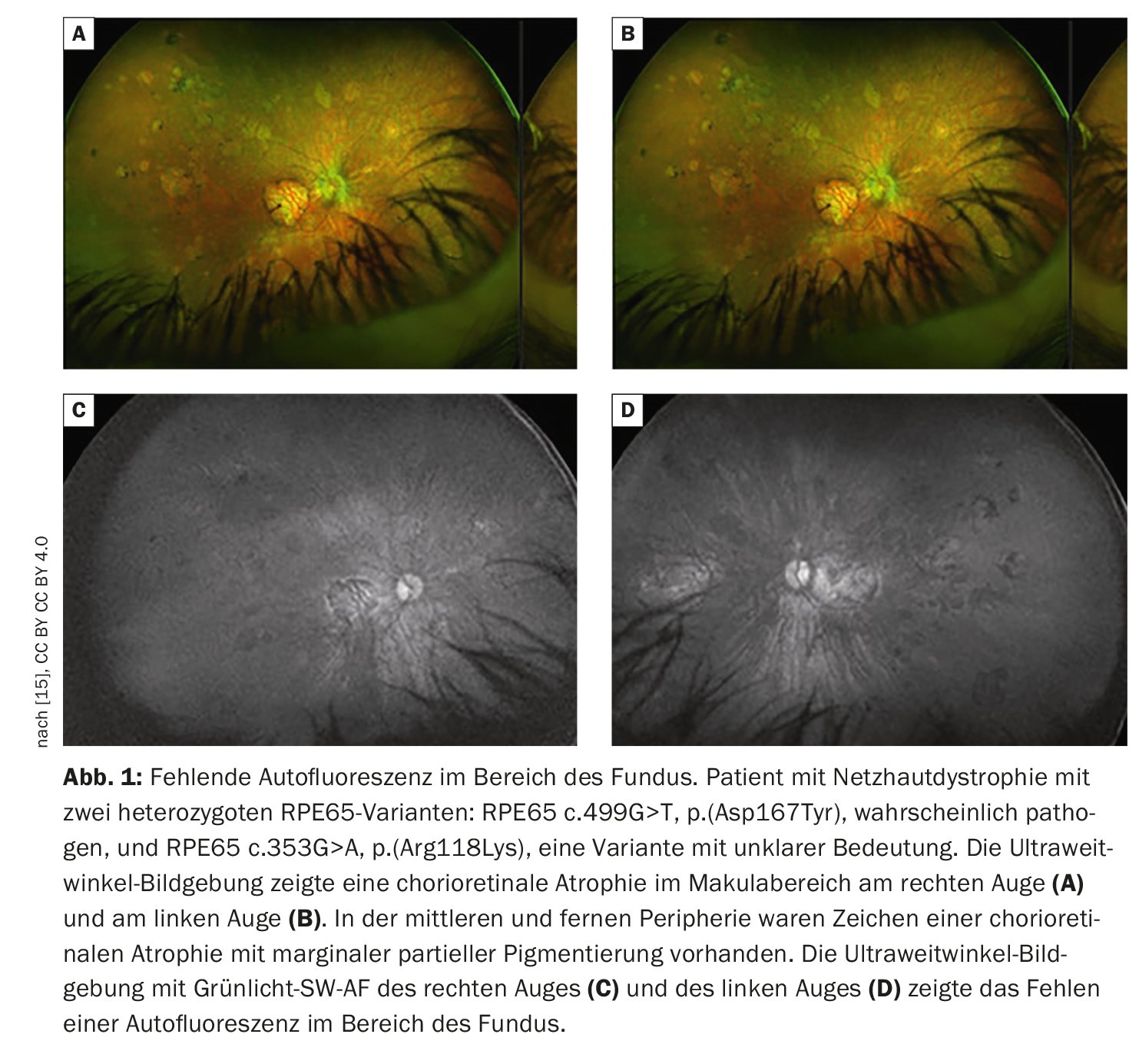
Retinal imaging provides more detailed visualization of retinal pathology than funduscopic examination. In addition to conventional color fundus photography, established techniques include high-resolution optical coherence tomography (OCT) and fundus autofluorescence, which can image fluorophores of the ocular fundus using short-wavelength light. Fundus autofluorescence involves imaging the distribution of fluorophores of the fundus of the eye, usually using short-wavelength excitation light in the blue or green range. In patients with mutations in the RPE65 gene, absent or at least reduced autofluorescence in the fundus region (Fig. 1) is a clinically significant feature due to defective metabolism of retinoids in photoreceptors and RPE cells [9]. For example, in retinitis pigmentosa, there is typically a concentric ring of increased autofluorescence for which there is no visible correlate funduscopically [10–12]. While the exact origin of this phenomenon is as yet incompletely understood, OCT studies have shown that the ring corresponds to the loss of the ellipsoidal band and a severe thinning or even loss of the photoreceptor layer [10].
Molecular genetic testing for detection of genetic cause
Phenotypes in variants in different genes often overlap and considerable variability is possible in variants in one gene. Therefore, molecular genetic testing is essential and an important basis for selecting the appropriate therapeutic option [6]. In 55-80% of all cases, the genetic cause of inherited retinal dystrophies can be identified by molecular genetic testing [4]. In patients with biallelic mutations in the RPE65 gene, RPE65 isomerase is either not expressed at all or expressed as a nonfunctional protein in the retinal pigment epithelium (RPE). By definition, for the diagnosis of biallelic RPE65 mutation-associated retinal dystrophy, both copies of the RPE65 gene must be present as either one or two distinct pathogenic variants [8]. The clinical phenotype in which biallelic RPE65 mutations manifest is usually associated with Leber congenital amaurosis or retinitis pigmentosa. Deficiency of functional RPE65 isomerase results in failure to convert the all-trans-retinal produced in the phototransduction cascade to 11-cis-retinal, which impairs regeneration of the visual pigment rhodopsin [1]. This disturbance in retinal metabolism causes photoreceptors to lose their ability to respond to light stimuli. Furthermore, accumulation of toxic intermediates leads to death of retinal pigment epithelial cells, resulting in additional photoreceptor dysfunction. This effect is more pronounced in photoreceptor rods than in photoreceptor cones, because the latter still have an alternative metabolic pathway for the regeneration of the visual pigment via the so-called Müller cells [1].
Gene therapy agent as a treatment option for mutation in the RPE65 gene.
The basic idea is that by adding a correct copy using retinal gene therapy, retinal function can be restored. Recombinant adeno-associated viral vectors (AAV) are well suited to perform gene transfer because they allow long-lasting and cell type-specific expression in the retina [9]. Voretigen Neparvovec is a gene transfer vector that uses the capsid of an adeno-associated serotype 2 (AAV2) viral vector as a transport vehicle for the cDNA of human retinal pigment epithelium-specific 65 kDa protein (hRPE65) into the retina [13].
| Voretigene neparvovec in biallelic RPE65 mutation. The efficacy of Luxturna® (voretigen neparvovec) was evaluated in a total of 41 patients, all of whom had hereditary retinal dystrophy [13]. In the main study, 11 adults (36%) and 20 children over 4 years of age (64%) participated. The average age was 15 years. To demonstrate the efficacy of Luxturna® in the main study, functional vision was measured. This is composed of visual acuity, visual field, and low-light perception and/or vision. After one year of treatment, patients in the Luxturna® treatment arm were able to complete a course more accurately, faster, and in lower light conditions. Improvement in functional vision was maintained over a period of at least three years. A 15-year follow-up of the 41 study participants is underway. |
Luxturna® is a gene therapy drug containing the active ingredient voretigen neparvovec, approved in Switzerland since February 2020 for the treatment of adults and children with vision loss due to hereditary retinal dystrophy with biallelic RPE65 mutation (box) [13]. Treatment with Luxturna® requires that the RPE65 mutation has been confirmed by genetic testing and that sufficient functional cells are still present in the patient’s retina [13]. Luxturna® is applied microsurgically under the retina [13,14]. In Switzerland, an exclusive center for this gene therapy is located at the University Hospital Basel (USB) [14]. Prof. Dr. med. Hendrik Scholl is Head and Chief Physician of the Eye Clinic at the USB, Prof. Dr. med. Christian Prünte is Clinical Chief Physician there and specializes, among other things, in microsurgical interventions. In November 2022, the USB announced that a twelve-member team led by Prof. Scholl and Prof. Prünte had performed the first microsurgical operation in Switzerland for the application of Luxturna®. The 51-year-old patient with hereditary retinal dystrophy underwent treatment of the second eye in March 2022, following surgery on the other eye a year earlier [14]. Both interventions went optimally, he said.
Take-Home Messages
- To date, more than 250 mutations are known to underlie hereditary retinal dystrophy. In 55-80% of all cases, the genetic cause can be identified by molecular genetic testing [4].
- The underlying genetic defect is defining for the molecular pathogenesis, the clinical picture and the course of the disease. Variants of the RPE65 gene are the cause of 0.6-6% of all cases of retinitis pigmentosa and 3-16% of cases of Leber congenital amaurosis/retinal dystrophy with early onset [2].
- In retinal imaging, mutations in the RPE65 gene are characterized by absent or reduced autofluorescence in the fundus.
- Luxturna® (voretigene neparvovec) is the first gene therapy available for the treatment of hereditary retinal dystrophy with biallelic RPE65 mutation [13].
Literature:
- Dias MF, et al: Molecular genetics and emerging therapies for retinitis pigmentosa: Basic research and clinical perspectives. Progress in Retinal and Eye Research 2017 (63): 107-131.
- Aoun M, et al: Hereditary retinal dystrophies due to RPE65 variants: From genetic diagnosis to therapy. Compass Ophthalmol 2021; 7: 115-123.
- Nash BM, et al: Retinal dystrophies, genomic applications in diagnosis and prospects for therapy. Translational Pediatrics 2015; 4 (2): 139-163.
- Kohl S, Biskup S: Genetic Diagnostic Testing in Inherited Retinal Dystrophies. Clin Monbl Ophthalmology 2013; 230(3): 243-246.
- Bolz HJ: Diagnostic analyses of retinal dystrophy genes: current status and perspective. Clin Monbl Ophthalmology 2021; 238: 261-266.
- Kellner U, et al: Diagnosis of hereditary retinal dystrophies. Value of molecular genetic diagnostics from the patient’s perspective. Ophthalmology 2022; 119: 820-826.
- Dockery A, et al: Next-generation sequencing applications for inherited retinal diseases. Int J Mol Sci 2021; 22: 5684.
- Blue Cross and Blue Shield of Kansas City (BCBSKC) 2018. Gene Therapy for Inherited Retinal Dystrophy: Policy Number 2.04.144.
- Stieger K, Lorenz B: Gene therapy for degenerative retinal diseases, www.kaden-verlag.de/fileadmin/images/ZPA_direkt/CME_ZPA_12-09.pdf,(last accessed 04.04.2023).
- Birtel J, et al: Diagnosis of hereditary retinal diseases Klin Monatsbl Augenheilkd 2021; 238: 249-260.
- Robson AG, et al: Serial imaging and structure-function correlates of high-density rings of fundus autofluorescence in retinitis pigmentosa. Retina 2011; 31: 1670-1679.
- Lima LH, et al: Progressive constriction of the hyperautofluorescent ring in retinitis pigmentosa. Am J Ophthalmol 2012; 153: 718-727.
- Drug Information, www.swissmedicinfo.ch,(last accessed 04.04.2023).
- “First use of gene therapy against blindness in Switzerland,” www.unispital-basel.ch/newscenter/zuweisenden-blog/15-07-2022,(last accessed 04.04.2023).
- Bjeloš M, et al: RPE65 c.353G>A, p.(Arg118Lys): A Novel Point Mutation Associated with Retinitis Pigmentosa and Macular Atrophy. International Journal of Molecular Sciences. 2022; 23(18):10513. www.mdpi.com/1422-0067/23/18/10513,(last accessed 04.04.2023).
HAUSARZT PRAXIS 2023; 18(4): 46-47