Acute infections activate various inflammatory and procoagulant mechanisms and are considered transient risk factors for venous thromboembolism (VTE). Infections not associated with bed rest and hospitalization also increase the risk of VTE. The cause-and-effect structure is complex. To assess the risk of thrombosis in infectious patients, one can use the “red flags” as a guide. Regarding infectious patients with atrial fibrillation, anticoagulation is indicated if there is no specific bleeding tendency.
According to current knowledge, infections are associated with a comparable increase in risk as the traditional risk factors, which are known to include major surgery within the last three months, immobilization lasting at least three days, or active tumor disease in the last six months, explains Prof. Philip Tarr, MD, Kantonsspital Baselland, Bruderholz [1]. Although infections have not been included in diagnostic scores to determine clinical pretest probability, their association with venous thromboembolism (VTE) is well documented in the literature [1].
Increased risk of VTE after respiratory or urinary tract infections.
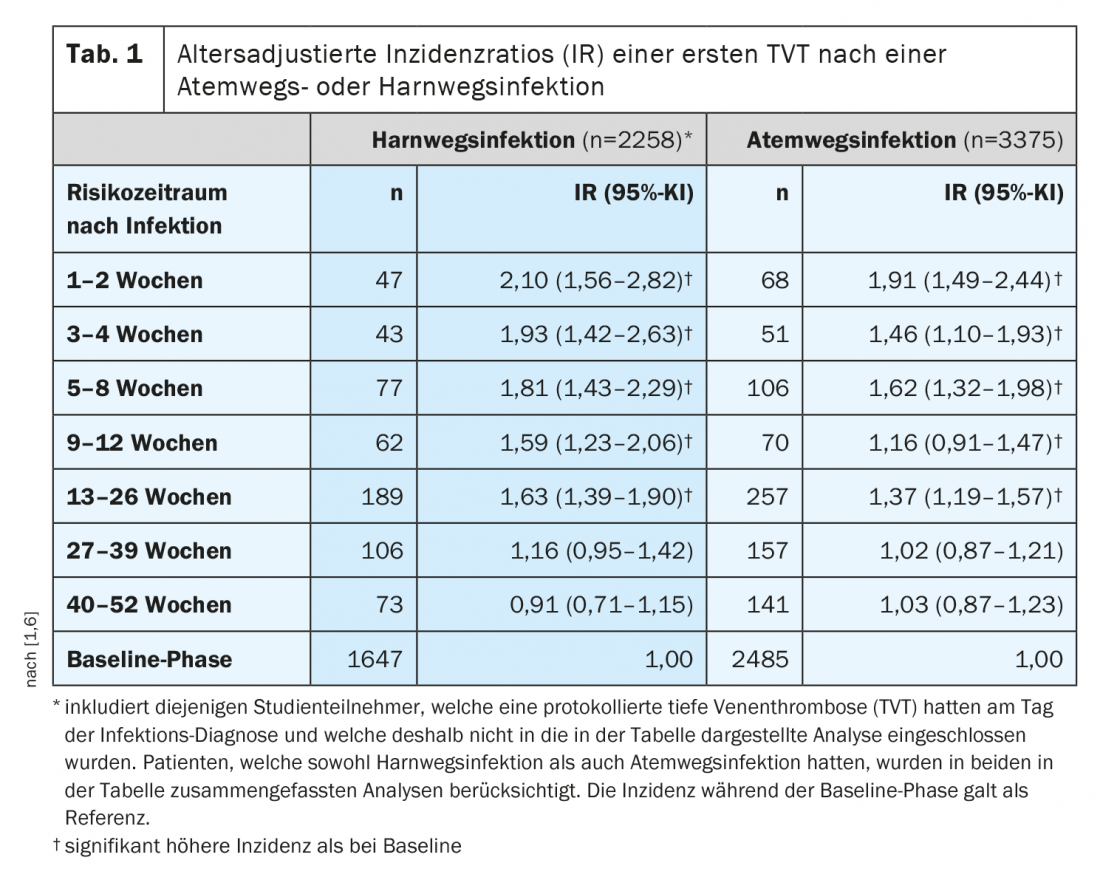
Hospitalized patients with infection have approximately twice the risk of VTE compared with hospitalized patients without infection [1]. Even banal infections without bedriddenness and hospitalization increase the risk of VTE, such as respiratory infections and urinary tract infections, which affect several hundred thousand people in Switzerland each year. “The risk of thromboembolism is highest in the first 1-2 weeks after an infection, but – and this is exciting – the increased risk of thrombosis persists for several months, up to half a year after an infection – at least statistically,” the speaker said. A 2- to 5-fold increased risk of VTE is thought to persist approximately 2 to 4 weeks after a simple respiratory or urinary tract infection [1,6]. Although this risk decreases over time, it remains elevated for 6-12 months after infection in both hospitalized and non-hospitalized patients according to scientific analyses (Table 1) [1].
Recognizing warning signs: which infectious patients are particularly at risk?
An association between inflammation and thrombosis propensity seems plausible, but the role of CRP has not been fully elucidated (Box) [2]. With regard to D-dimer, Prof. Tarr advises against routine determination on the grounds that elevation is often present. However, a normal D-dimer was a good argument against DVT/LE [1]. The speaker summarized the “red flags” regarding deep vein thrombosis/pulmonary embolism in infectious patients as follows [1]:
- Dyspnea/thoracic pain
- Circumferential difference extremity (also arm in case of central venous catheter, port, picc line)
- If the clinical course is more consistent with DVT/LE and not with (suspected) infection
- If the fever does not subside, despite adequate antibiotic therapy.
- In case of fever without apparent cause
- Erysipelas: in 2-3% of cases DVT is present

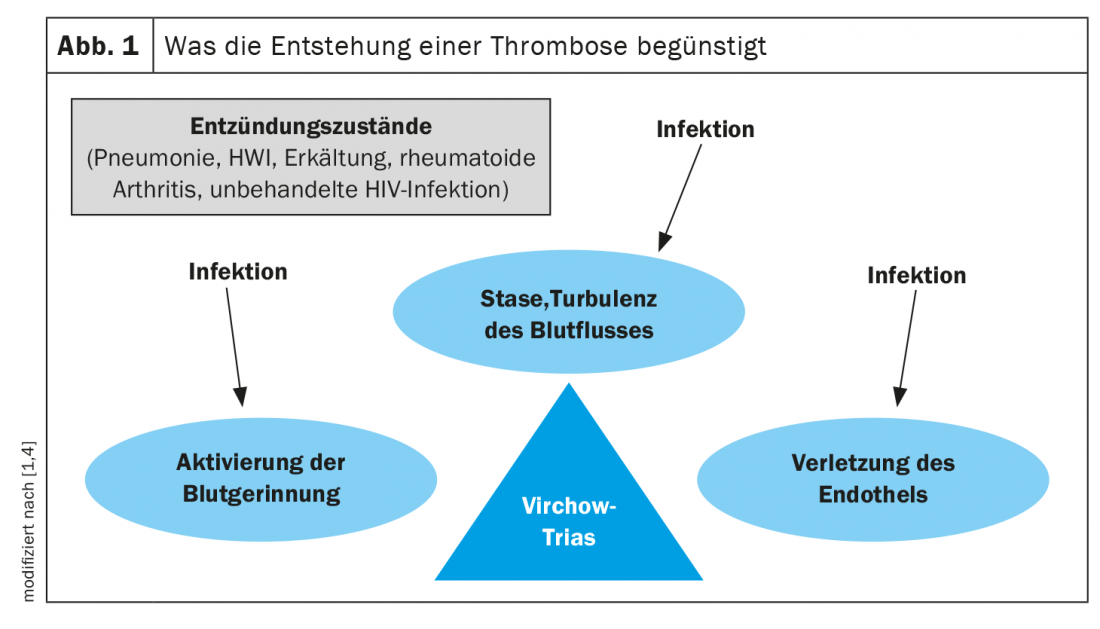
Pathomechanisms: Virchow triad
The cause-and-effect relationship between infection and thrombosis is extremely complex. Acute infection is known to activate various inflammatory and procoagulant mechanisms [1–3]. Rudolf Virchow’s famous triad states that thrombosis is favored by three factors: Activation of blood coagulation (hypercoagulability), venous stasis, and endothelial injury (Fig. 1) [1,2,4]. These mechanisms are influenced by infections. Thus, an inflammatory state activates various acute-phase proteins of the coagulation cascade, which have procoagulant effects and thus increase the risk of thrombosis. These include, for example, CRP, fibrinogen, factor VIII, interleukin-6, and von Willebrand factor (vWF) [1,2]. It is not fully understood whether infection-related, local or systemic inflammation plays the key role in this process and whether specific bacteria (e.g., S. aureus or gram-negative rods) particularly increase the risk of VTE [2,5]. Antiphospholipid antibodies (aPL) are also associated with the occurrence of thrombosis, which are rarely triggered by infections and more frequently by autoimmune diseases such as lupus erythematosus and are a trigger of the so-called antiphospholipid syndrome (APS) [2]. Moreover, certain bacterial toxins and viral infections such as SARS-CoV-2, cytomegalovirus, or Epstein-Barr virus can damage the endothelium [2]. VTE risk factors such as immobilization, age, tumor disease, or immunosuppression further increase the risk for infection [2].
Anticoagulation in infectious patients with atrial fibrillation.
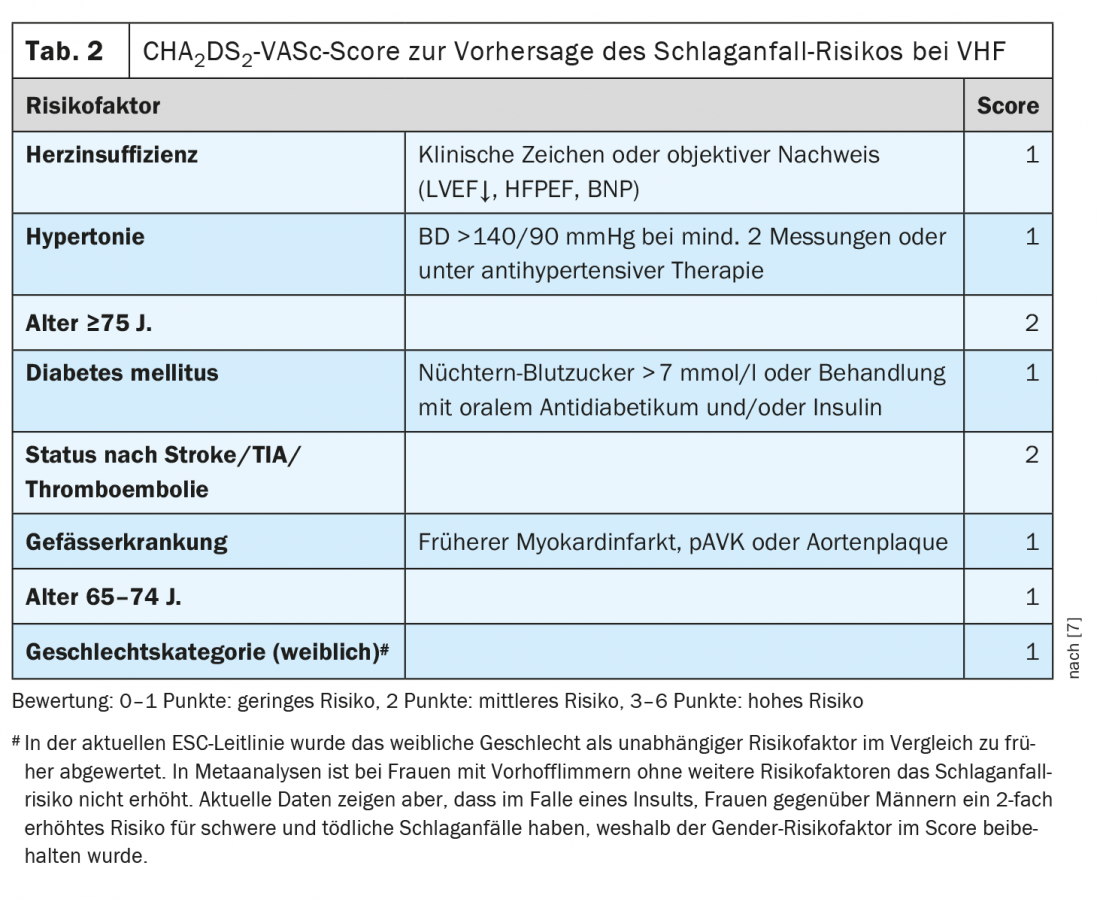
“Pneumonia is associated with a cardiovascular event in approximately 1 in 5 patients,” emphasized the speaker [1]. This may be an (acute) coronary syndrome or myocardial infarction, but it may also be a new onset arrhythmia (especially atrial fibrillation) or decompensation in known heart failure. “Pneumonia leads to procoagulant mechanisms that are activated by inflammation,” elaborates Prof. Tarr [1]. Cardiovascular events in pneumonia are often missed and are associated with increased mortality. The risk of acute coronary syndrome (ACS) increases proportionally with increasing severity of pneumonia, but is increased even with a mild course of pneumonia over several weeks to months. The association with ACS has been documented not only for pneumonia but also for influenza, urinary tract infections, and bacteremia. Infections can also trigger atrial fibrillation. This increases the risk of embolic stroke. This has therapeutic implications. “If a patient goes into atrial fibrillation in the setting of pneumonia, is that now provoked atrial fibrillation that you only need to anticoagulate for a short period of time?” From today’s perspective, the answer is “unfortunately no,” says Prof. Tarr. “We are supposed to discount the infection as the trigger of atrial fibrillation,” the speaker adds, adding, “The fact that atrial fibrillation has occurred at all due to an infectious trigger indicates that the atrium is diseased/dilated. In the context of infection, there may be inflammation and electrolyte shifts, altered parasympathetic or sympathetic activity, which ultimately triggered the AF” [1]. Depending on the CHA2DS2-VASc score, the ESC and AHA guidelines recommend lifelong anticoagulation if AF occurs (Table 2) [1,7–9]. Apparently, there is a predisposition to atrial fibrillation and, according to current knowledge, the embolic stroke risk in paroxysmal infarct-triggered atrial fibrillation is comparable to that in permanent atrial fibrillation [1].
Source: Medical Congress Davos
Literature:
- Tarr P: Infection and thrombosis: Folie à deux. Prof. Philip Tarr, MD. Medical Congress Davos, 10.-11.02.2022
- Pfister T, et al: Infection and thrombosis, part 1: background. Prim Hosp Care Allg Inn Med 2021; 21(04): 125-130.
- Riva N, Donadini MP, Ageno W: Epidemiology and pathophysiology of venous thromboembolism: similarities with atherothrombosis and the role of inflammation. Thromb Haemost 2015; 113(6): 1176-1183.
- Bagot CN, Arya R: Virchow and his triad: a question of attribution. British Journal of Haematology 2008; 143(2): 180-190.
- White RH: Identifying Risk Factors for Venous Thromboembolism. Circulation 2012; 125(17): 2051-2053.
- Smeeth L, et al: Risk of deep vein thrombosis and pulmonary embolism after acute infection in a community setting. Lancet 2006; 367(9516): 1075-1079.
- Rosemann A: Atrial Fibrillation, 11/2018, www.medix.ch/media/gl_vorhofflimmern_2018_31.10.19_mh.pdf, (last accessed Mar 28, 2022).
- Kirchhof P, et al: European Heart Journal. 2016 Oct 7;37(38): 2893-2962.
- January CT, et al: Circulation 2019 Jul 9; 140(2).
HAUSARZT PRAXIS 2022; 17(4): 36-37 (published 6.4.22, ahead of print).