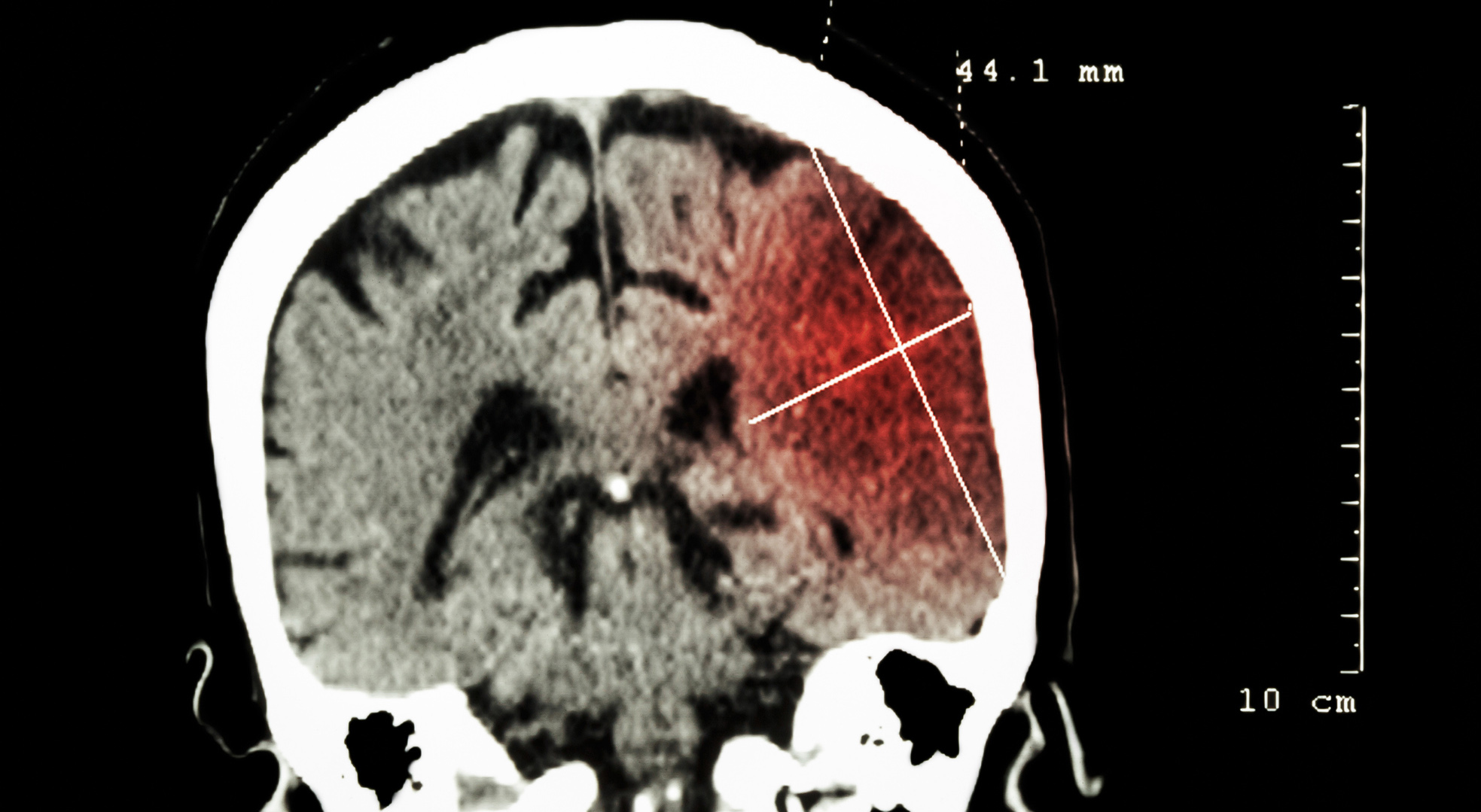
The patient, who had a phenotype characterized by hypertelorism, thick eyebrows and other features, was found to have a c.517C>T nonsense mutation in the KMT2A gene. This rare heterozygous mutation leads to a premature stop codon and haploinsufficiency. As the mutation was not detectable in the parents, it arose de novo in the patient. This case report emphasizes the importance of exome sequencing in the diagnosis of chromatinopathies, especially when clinical features overlap with other syndromes.
Publikation
- HAUSARZT PRAXIS